As doenças do músculo e da junção neuromuscular constituem um grupo heterogêneo de distúrbios adquiridos e hereditários. Quando se manifestam, os sintomas são variados e incluem a fadigabilidade, enfraquecimento da musculatura esquelética, atrofia, cãibras musculares ou mialgias e comprometimento da função dos músculos respiratórios, faríngeos, faciais e oculares. Em geral, os músculos proximais são mais seletiva e severamente afetados do que os músculos distais. Os pacientes costumam relatar uma dificuldade crescente para executar as tarefas diárias que requerem predominantemente o uso da musculatura proximal, como levantar-se de uma cadeira, subir escadas, subir no meio-fio, levantar objetos ou pentear o cabelo. Os movimentos motores precisos, que dependem da força dos músculos distais, como abotoar uma camisa, agarrar objetos, um aperto de mãos ou elevar pés e dedos do pé, são afetados em fases mais tardias do curso da doença, porém mais cedo nas miopatias de aparecimento distal. O envolvimento dos músculos oculares, que se manifesta como ptose, diplopia ou limitação dos movimentos oculares, direciona a atenção para certos tipos de miopatias congênitas ou adquiridas e para os distúrbios da junção neuromuscular. Isto também é válido para o envolvimento da musculatura facial, faríngea e cervical ou axial, com resultante diminuição da expressão facial, disfonia, disfagia, dificuldade para sustentar a cabeça ereta ou camptocormia. Os músculos respiratórios também podem ser afetados, em geral nos estágios mais avançados, porém relativamente mais cedo em determinadas condições. O enfraquecimento crônico quase sempre está associado ao desgaste muscular. Os reflexos tendíneos costumam ser preservados, mas podem se tornar ausentes em músculos severamente enfraquecidos ou atrofiados. A maioria das miopatias afeta somente a musculatura esquelética, entretanto em alguns casos também pode haver comprometimento da musculatura lisa e cardíaca. Os pacientes com miopatia não apresentam distúrbios sensoriais nem disfunção autonômica, porque os nervos periféricos e o sistema nervoso autônomo são preservados.
O objetivo do exame clínico é encontrar achados junto aos músculos e excluir doenças capazes de mimetizar os sinais e sintomas miopáticos, como as síndromes neuronais motoras, neuropatias motoras ou doenças psicogênicas. A obtenção de uma história familiar completa e o exame dos familiares do paciente muitas vezes são medidas necessárias à exclusão da hipótese de uma doença hereditária. Os marcadores de autoimunidade, como diversos autoanticorpos, paraproteínas, ligações imunogenéticas ou coexistência com outros distúrbios autoimunes, são úteis para excluir as miopatias imunomediadas potencialmente tratáveis. As avaliações laboratoriais úteis realizadas de modo seletivo, de acordo com a apresentação clínica, incluem (1) exames para exclusão de uma doença sistêmica, fatores exógenos, toxinas ou vírus que possam induzir miopatia; (2) exames eletrofisiológicos para localização da lesão junto ao músculo ou na junção neuromuscular e para excluir distúrbios de nervo periférico ou neuronais motores; (3) determinação dos níveis séricos de enzimas musculares; (4) quantificação de autoanticorpos específicos associados aos antígenos presentes no músculo ou na junção neuromuscular; (5) biópsia de músculo para realização de exames de histoquímica enzimática, imunocitoquímica, microscopia eletrônica, quantificação bioquímica de uma proteína ou enzima muscular específica, além de alguns testes genéticos; (6) teste do exercício para medir a produção de lactato e amônia, diante da suspeita de miopatia metabólica; (7) testes genéticos sobre linfócitos do sangue periférico, caso o gene seja conhecido; e (8) exames de imagem do músculo, que são determinados pelo problema clínico específico investigado.
A principal tarefa do médico é identificar quais miopatias são tratáveis, a fim de iniciar a terapia sem demora, antes que haja desenvolvimento de um enfraquecimento permanente. Para os pacientes com distúrbios intratáveis, é essencial fornecer terapia de suporte, reabilitação, aconselhamento genético e apoio psicológico.
Neste capítulo, são descritas as miopatias e distúrbios de junção neuromuscular mais comuns, enfatizando-se o quadro clínico, a patogênese, o diagnóstico e a terapia. A esclerose lateral amiotrófica (ELA), que é semelhante a certas miopatias, é brevemente discutida devido ao caráter essencial da detecção precoce, aconselhamento adequado, possível participação em estudos clínicos e terapia de suporte. O grupo das miopatias inflamatórias, que é abordado em outro texto, não será discutido aqui.
Distrofias musculares
As distrofias musculares constituem um grupo heterogêneo de doenças musculares congênitas caracterizadas por enfraquecimento muscular severo, atrofia, elevação dos níveis séricos de enzimas musculares e alterações citoarquitetônicas destrutivas das fibras musculares. A classificação tradicional das distrofias musculares em Duchenne (DMD), Becker (DMB) e das cinturas pélvica e escapular (DMCPE) mudou, pois foram identificados defeitos genéticos envolvendo proteínas musculares responsáveis pela maioria destas doenças e foi demonstrado que a deficiência de proteínas musculares específicas atua como causa destas condições.
Exames musculares, bioquímicos e imunocitoquímicos identificaram a distrofina-glicoproteína como sendo um complexo de múltiplas subunidades proteicas atuante na ligação do citoesqueleto à matriz extracelular1-3 [Figura 1]. As deficiências de certos componentes deste sistema acarretam instabilidade do sarcolema, resultando em necrose da fibra muscular e desenvolvimento de síndromes clínicas específicas.1-3 As distrofias musculares atualmente são mais bem classificadas de acordo com o gene e a proteína defeituosa envolvida. Esta proteína defeituosa pode ser um componente do núcleo, do citosol, do citoesqueleto, do sarcolema, da matriz extracelular ou do filamento intermediário. As distrofias musculares mais comuns são listadas na Tabela 1. O papel de cada proteína na sustentação, reforço ou conexão do núcleo com o citoesqueleto, sarcolema e matriz extracelular [Figura 1] é discutido nas seções que descrevem cada distúrbio específico.

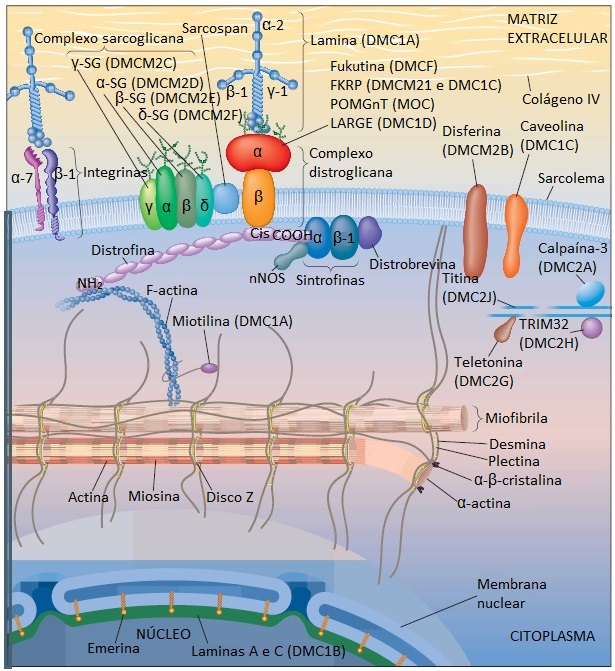
Figura 1. Conceito em vigor da organização molecular do complexo distroglicana no sarcolema extrajuncional. A deficiência ou ausência de várias proteínas resulta em distrofias musculares particulares. As mutações nestes genes determinantes de distrofia produzem distrofia muscular de Duchenne (DMD) ou distrofia muscular de Becker (DMB). As mutações nos genes determinantes do complexo sarcoglicana produzem vários subtipos de distrofias musculares das cinturas pélvica e escapular (DMCPE). Por fim, as mutações no gene codificador de alfa-2-lamina (merosina) produzem distrofia muscular congênita (DMC). Outras proteínas localizadas no complexo distrofina-glicoproteína incluem a caveolina, óxido nítrico sintase neuronal (nNOS), distrobrevina, sintrofinas e actina. O colágeno VI é um componente da lâmina basal.
Cis = domínio rico em cisteína; COOH = carboxiterminal; FKRP = proteína relacionada à fukutina; NH2 = aminoterminal.
Tabela 1. Distrofias musculares com suas respectivas localizações genéticas e produtos
Doença
|
Locus do gene
|
Produto do gene
|
Distúrbios alélicos
|
Distrofias musculares recessivas ligadas ao X
| |||
DMD
|
Xp21
|
Distrofina
|
Miocardiopatia isolada, DMB
|
DMB
|
Xp21
|
Distrofina
|
Miocardiopatia isolada, DMD
|
DMED
|
Xq28
|
Emerina
|
DMCM1B
|
DMCPE autossômica dominante
| |||
DMCPE1A
|
5q22-5q31
|
Miotilina
|
MMF
|
DMCPE1B
|
1q11-21
|
Laminas A e C
|
DMED autossômica dominante
|
DMCPE1C
|
3p25
|
Caveolina-3
|
Doença do músculo ondulado; hiper-CKmia
|
DMCPED
|
7q
|
Desconhecido
| |
DMCPEE
|
7q
|
Desconhecido
|
Miocardiopatia e defeito de condução
|
DMCPE autossômica recessiva
| |||
DMCPE2A
|
15q15
|
Calpaína-3
| |
DMCPE2B
|
2p13
|
Disferlina
| |
DMCPE2C
|
13q12
|
Gamassarcoglicana
| |
DMCPE2D
|
17q12-q21
|
Alfassarcoglicana
| |
DMCPE2E
|
4q12
|
betassarcoglicana
| |
DMCPE2F
|
5q33-q34
|
deltassarcoglicana
| |
DMCPE2G
|
17q11-q12
|
Teletonina
| |
DMCPE2H
|
9q3-q34
|
TRIM32
| |
DMCPE2I
|
19q13.3
|
FKRP
|
DMC1C, DMCF
|
DMCPE2J
|
2q24.3
|
Titina
| |
DMC
| |||
DMCIA “clássica” DMC
|
6q22
|
Alfa-2-lamina (merosina)
| |
DMC1B
|
12q13
|
Alfa-7-integrina
| |
DMC1C
|
19q13.3
|
FKRP
|
DMCM2I
|
DMC1D
|
LARGE
| ||
DMCF
|
9q31-q33
|
Fukutina
| |
MOC
|
1p32-p34
|
POMGnT
| |
Síndrome de Walker-Warburg
|
?
|
POMGnT
| |
Outras DMC
| |||
DMC com miopatia da espinha rígida
|
1q35-36
|
Selenoproteína N
| |
Miopatia autossômica dominante de Betlem
|
21q22
|
Colágeno VI alfa-1, beta-2
| |
Miopatia de Ullrich
|
2q37
|
Colágeno VI alfa-3
| |
Miopatia do filamento intermediário
| |||
Desmina
|
2
|
Desmina
| |
Alfa-beta-cristalina
|
11q21-23
|
Desmina, alfa-beta-cristalina
| |
Epidermólise bolhosa e distrofia muscular
|
8q24-qter
|
Plectina
| |
Miotilina
|
5q22-5q31
|
Miotilina
| |
Distrofias autossômicas dominantes com fenótipo único
| |||
Distrofia miotônica
| |||
DM1
|
19q13
|
Miotonina-proteína quinase
| |
DM2
|
3q21
|
Proteína em dedo de zinco 9
| |
Atrofia muscular facioscapuloumeral
|
4q35
| ||
DMOF
|
14q11.2-q13
|
Proteína ligadora de poliA 2
|
DMB = distrofia muscular de Becker; DMC = distrofia muscular congênita; DMCF = distrofia muscular congênita de Fukuyama; DMCPE = distrofia muscular da cintura escapular/pélvica; DMD = distrofia muscular de Duchenne; DMED = distrofia muscular de Emery-Dreifuss; DMOF = distrofia muscular oculofaríngea; FKRP = proteína relacionada à fukutina; MMF = miopatia miofibrilar; MOC = doença do músculo-olho-cérebro.
Distrofias musculares recessivas ligadas ao X
Distrofinopatias
As distrofinopatias são causadas pela deficiência de distrofina, uma proteína de 427 kDa que pertence ao citoesqueleto e possui formato de bastão. A distrofina constitui 5% de todas as proteínas citoesqueléticas do sarcolema e atua ancorando a F-actina (a forma filamentar da actina) na membrana plasmática (sarcolema) do músculo [Figura 1].2-7 A distrofina parece reforçar e estabilizar a membrana plasmática durante o estresse produzido pela contração muscular, mantendo a ligação mecânica entre o citoesqueleto e a matriz extracelular. A deficiência ou ausência de distrofina estão associadas a várias distrofinopatias, das quais o protótipo é a DMD.4-7
Distrofia muscular de Duchenne (DMD). A DMD é um distúrbio recessivo ligado ao X [Tabela 1] produzido por mutações no gene codificador da distrofina, que está localizado no braço curto do cromossomo X, na posição Xp21. O gene da distrofina contém mais de 2.000 kb de DNA. Em 65 a 70% dos casos, a DMD resulta de amplas deleções (vários kb) neste gene e da consequente falta de distrofina muscular. As mutações espontâneas também são encontradas com frequência em pacientes com DMD.4-7 A ausência da distrofina resulta no enfraquecimento e na quebra da membrana do sarcolema, permitindo a entrada de cálcio e consequente necrose da fibra muscular. As deleções, detectadas no DNA extraído dos linfócitos do sangue periférico, rompem a estrutura de leitura aberta dos códons em trinca do RNA mensageiro (mRNA) e produzem as formas severas de DMD. As duplicações genéticas parciais são responsáveis por 6% das mutações envolvendo a distrofina.
O teste da reação em cadeia da polimerase (PCR), que investiga os conhecidos hot spots (manchas quentes) em dois éxons, detecta quase 2/3 dos casos de DMD varrendo o DNA oriundo do sangue. Entretanto, esta técnica não detecta as mutações pequenas (p. ex., mutações pontuais e erros de splicing) que produzem uma proteína de distrofina truncada e são responsáveis por até 30% dos casos de DMD. Um método altamente sensível, baseado no polimorfismo conformacional de fita única, varre os 79 éxons do gene da distrofina e detecta 90% das mutações de DMD por análise de DNA obtido do sangue periférico.7 Resultados similares são fornecidos pela PCR seguida da análise de sequência direta (isto é, sequenciamento com primer/interno de amplificação de condição única [SCAIP – em inglês, single condition amplification internal/primer sequencing]).8
A DMD ocorre em 1 a cada 3.000 nascimentos de bebês do sexo masculino. Os meninos afetados tornam-se sintomáticos depois que começam a andar, geralmente aos 2 a 3 anos de idade. A ocorrência de DMD em meninas é extremamente rara [ver Portadoras do sexo feminino e distrofinopatias em mulheres, adiante]. As manifestações iniciais são o andar desajeitado, postura lordótica, hipertrofia da panturrilha, contraturas articulares e andar na ponta dos dedos. A estas manifestações segue-se um progressivo enfraquecimento e desgaste muscular. Torna-se difícil realizar os movimentos de levantar-se do chão ou de uma cadeira baixa, subir escadas e erguer o braço. O esvaziamento gástrico retardado pode causar episódios repentinos de vômito e dor abdominal. A doença não passa e aumenta progressivamente de intensidade. As crianças afetadas são dependentes de cadeiras de rodas ao completarem 12 anos de idade e, quando atingem 25 anos, a maioria morre em decorrência das complicações da insuficiência respiratória. Embora a DMD seja uma doença da musculatura esquelética, é comum o acometimento do miocárdio, e há desenvolvimento de insuficiência cardíaca congestiva e arritmia durante a fase mais tardia da doença.9 Também há um envolvimento leve e não progressivo do sistema nervoso central (SNC), que se manifesta como irritabilidade, hiperatividade ou disfunção cognitiva.
Os exames laboratoriais de rotina dos pacientes com DMD a princípio mostram níveis séricos de creatinina quinase (CK) de até 20.000 UI/L, porém esta concentração declina de modo estável conforme a massa muscular vai sendo depletada. As biópsias de músculo revelam a existência de severa miopatia destrutiva. Algumas células T CD8+ frequentemente estão presentes e invadem as fibras musculares. Numerosos macrófagos estão associados à fagocitose das fibras necróticas. Observa-se aumento da concentração de tecido conectivo e a presença de fibras musculares hipercontraídas é comum. O diagnóstico é confirmado pela ausência da distrofina, que é demonstrada pela análise imunocitoquímica de cortes de biópsia de músculo ou por immunoblots preparados com amostras de biópsia de músculo coradas com anticorpos antidistrofina.10
O tratamento é totalmente sintomático, enfatizando a instituição de terapia respiratória sistemática e fisioterapia, bem como o fornecimento de suporte psicossocial ao paciente e seus familiares. O aconselhamento genético é altamente apropriado. A presença de inflamação endomisial leva à pronta instituição do tratamento da DMD com glicocorticoides. Em um estudo controlado, o uso de esteroides resultou em uma melhora discreta e temporária, além de ter retardado a evolução da doença.10 O uso prolongado de esteroides é limitado por efeitos colaterais severos, em especial a obesidade, fraturas, osteoporose, diabetes e hipertensão. O deflazacorte, um esteroide que produz menos efeitos colaterais de mineralocorticoides, parece ser um pouco mais seguro.11 A suplementação com creatina e coenzima Q10 costuma ser utilizada.12 Em uma forma modificada de terapia genética, mioblastos humanos contendo distrofina normal foram injetados na musculatura de pacientes com DMD e falharam em promover qualquer tipo de melhora em termos de força. Um estudo com gentamicina, capaz de “ler do princípio ao fim” as mutações nonsense e gerar uma proteína de comprimento integral, restaurando assim a função da distrofina,13,14 foi igualmente decepcionante. É possível que as futuras terapias genéticas se mostrem eficazes quando forem descobertos vetores adequados para serem usados na inserção efetiva do gene no músculo.15
Distrofia muscular de Becker (DMB). A DMB e a DMD são distúrbios alélicos[Tabela 1], porém a DMB geralmente se manifesta mais tarde e apresenta evolução mais lenta.
Cerca de 65% dos pacientes com DMB apresentam deleções de estrutura no gene da distrofina, mas produzem uma proteína frequentemente truncada e apenas semifuncional.2-6,10 A imunocitoquímica do músculo de um paciente com DMB empregando anticorpos antidistrofina mostra uma coloração preservada e atenuada do sarcolema (em vez da ausência de coloração observada na DMD), revelando uma fragmentação da membrana nas áreas imunocoradas. O immunoblot detecta concentrações reduzidas de uma distrofina, cujo tamanho é menor ou maior que o tamanho da distrofina normal.
A idade do paciente no momento do aparecimento da DMB é variável. É possível identificar casos em pacientes com idades que variam de 3 a 70 anos, porém a média da idade dos pacientes no início da DMB é 12 anos. A expressão fenotípica da DMB também apresenta um amplo espectro. Os pacientes desenvolvem graus variáveis de enfraquecimento da musculatura proximal e níveis séricos de CK que chegam a 20 vezes os níveis normais. As formas brandas manifestam-se apenas como cãibras musculares, intolerância ao exercício, mioglobinúria, elevação assintomática dos níveis séricos de CK, enfraquecimento muscular leve ou miopatia do quadríceps.2-6 A dor na panturrilha ao exercício é constantemente um dos sintomas, e a ampliação da panturrilha também é comum. A maioria dos pacientes perde a capacidade de deambulação ao redor dos 40 anos de idade (entre 10 e 70 anos). A idade do paciente ao morrer varia de 23 a 89 anos (em média, 42 anos de idade). Os achados de biópsia muscular são semelhantes àqueles encontrados nos pacientes com DMD, porém menos severos. Em pacientes com menos de 8 anos de idade, a manifestação da DMB costuma ser indistinguível da manifestação da DMD. As manifestações cardíacas são comuns, e a miocardiopatia pode ser grave. A severidade dos sintomas cardíacos, porém, não está relacionada à severidade da miopatia. Não existe nenhum tratamento efetivo para a DMB.
Portadoras do sexo feminino e distrofinopatias em mulheres. Uma história detalhada e o exame clínico de portadores assintomáticos do sexo feminino podem revelar um enfraquecimento muscular leve, cãibras musculares, hipertrofia isolada da panturrilha, fadiga e níveis séricos elevados de CK.3-7 O exame da biópsia de músculo revela a presença de fibras negativas para distrofina. Em pacientes heterozigotos, quando é feita a amplificação dos éxons específicos com propensão a deleções situados no gene da distrofina, as deleções são identificadas como uma redução de 50% na intensidade da banda de DNA amplificada, em comparação à banda correspondente aos éxons do tipo selvagem.4-7 Este método detecta cerca de 98% das deleções. Contudo, existem casos em que a mãe de um menino afetado não carrega em seu sangue a mutação encontrada no filho. Estes casos, que estão ligados a mutações associadas à DMD recém-identificadas, representam até 20% dos novos casos de DMD e resultam de mosaicismo gonadal materno (ou seja, as mutações são encontradas somente nos oócitos).4-7 As mulheres com este tipo de mutação podem gerar bebês do sexo masculino afetados ou bebês do sexo feminino portadores. As filhas destas mulheres devem ser examinadas para identificação de portadoras. Entretanto, como a mutação ocorre nos oócitos, as irmãs da mulher que tenha a mutação podem não ter herdado as mutações de seus pais e não precisam ser examinadas como potenciais portadoras.4-7 A manifestação da DMD em indivíduos heterozigotos do sexo feminino ocorre quando o cromossomo X paterno normal, que abriga o gene da distrofina normal, é inativado em uma ampla proporção de células embrionárias (hipótese de Lyon). Nestas pacientes do sexo feminino, a doença pode ser tão severa quanto nos pacientes do sexo masculino.
Miocardiopatia dilatada ligada ao X. A miocardiopatia dilatada ligada ao X resulta da deficiência de distrofina no miocárdio, e não no músculo esquelético. Os pacientes apresentam um distúrbio cardíaco progressivo e desenvolvem insuficiência cardíaca congestiva na 2ª ou 3ª décadas da vida. As portadoras do sexo feminino que manifestam a doença desenvolvem uma miocardiopatia de aparecimento lento, que surge em torno da 5ª década da vida. Foi proposto que as deleções próximas ao éxon 1 do gene da distrofina, as quais afetam a expressão ou função da distrofina no miocárdio, são a causa da doença.4-7
Distrofia muscular de Emery-Dreifuss (DMED)
A distrofia muscular de Emery-Dreifuss (DMED) possui 2 formas genéticas: uma doença recessiva ligada ao X, que foi mapeada em Xq28 (DMED-LX); e uma forma autossômica dominante menos comum, mapeada em 1q11-q23 (DMED-AD). Ambas são clinicamente indistinguíveis. A DMED-LX é causada por uma mutação que afeta a proteína de membrana nuclear emerina [Figura 1];16 a DMED-AD é causada por mutações no gene lamina A/C, que codifica 2 proteínas das lâminas nucleares: lamina A e lamina C.16As mutações no gene lamina A/C são mais comumente causadoras de miocardiopatia e defeitos de condução.16 As proteínas integrais da membrana nuclear interagem de perto com as laminas nucleares, que são proteínas do filamento intermediário encontradas na face nuclear (lado interno) da membrana nuclear [Figura 1]. A emerina liga-se à lamina A, que é um dos produtos do gene laminina A/C.
Os pacientes com DMED apresentam uma tríade de sintomas: (1) distribuição umeroperoneal do envolvimento muscular, com desgaste proeminente e enfraquecimento dos músculos bíceps, tríceps, tibial anterior e peroneal, que evoluem lentamente segundo um padrão escapulopelvicoperoneal e, então, incluem o envolvimento muscular peitoral e pélvico; (2) desenvolvimento precoce de contrações junto aos cotovelos, flexores, tendão de Aquiles, pescoço e coluna vertebral, que podem anteceder o enfraquecimento muscular significativo; e (3) envolvimento cardíaco, que se manifesta como defeitos de condução com bradicardia e um intervalo PR prolongado. A paralisia atrial isolada é fortemente sugestiva de DMED.
As portadoras de DMED do sexo feminino geralmente não apresentam enfraquecimento muscular. Contudo, podem desenvolver bloqueio cardíaco.
A DMED surge durante as primeiras 2 décadas da vida. Seu diagnóstico é estabelecido com base na manifestação clínica, níveis de CK discretamente elevados e achados da biópsia de músculo, sendo confirmado por análise de mutação. As biópsias de músculo revelam aspectos miopáticos inespecíficos, porém a ausência da emerina – que é evidenciada pelo uso de anticorpos antiemerina na análise de imunocitoquímica ou no ensaio de Western blot realizados com amostras de biópsia de músculo – sustenta o diagnóstico. Os testes genéticos disponíveis atualmente identificam as pacientes portadoras que podem apresentar defeitos de condução cardíaca. O pronto reconhecimento e a colocação de um marca-passo podem prevenir a morte súbita ou os ataques de síncope.
Não há nenhum tratamento específico, porém a fisioterapia pode retardar o desenvolvimento das contrações.
Distrofias musculares da cintura pélvica e escapular (DMCPE)
As DMCPE constituem um grupo heterogêneo de distúrbios denominados autossômicos dominantes (DMCPE1A-F), autossômicos recessivos (DMCPE2A-J) ou congênitos (DMC – distrofias musculares congênitas) [Tabela 1].4,6,17-20
Distrofias musculares da cintura pélvica e escapular autossômicas dominantes (DMCPE1)
Trata-se de distúrbios geralmente incomuns, que representam menos de 10% de todas as DMCPE [Tabela 1]. Estes distúrbios manifestam-se como um enfraquecimento muscular proximal e distal de progressão lenta, e também pela elevação dos níveis séricos de CK. Tendem a ser mais brandos do que as outras formas de DMCPE. O diagnóstico é estabelecido pela identificação da proteína ausente e do defeito genético na análise de mutação. Não há tratamento específico para este grupo de distúrbios.
DMCPE1A. A DMCPE1A resulta de mutações que afetam a miotilina, uma proteína necessária à montagem normal e manutenção do sarcômero.21 Além do enfraquecimento muscular proximal e distal, os sintomas podem incluir disartria, hipofonia e voz nasalada. Este distúrbio é alélico para a forma de miopatia miofibrilar (MMF) causada pela mutação na miotilina [ver Miopatias miofibrilares (MMF) decorrentes de mutações em proteínas do filamento intermediário, adiante].21
DMCPE1B. A DMCPE1B é causada por mutações no gene lamina A/C. O fenótipo deste distúrbio é idêntico ao fenótipo da DMCPE-AD [ver Distrofia muscular de Emery-Dreifuss (DMED), anteriormente]. Além de apresentarem os sintomas de DMCPE, alguns pacientes desenvolvem uma lipodistrofia parcial familiar, que é caracterizada por diminuição do tecido adiposo subcutâneo, resistência à insulina, níveis aumentados de triglicerídeos, níveis baixos de lipoproteínas de alta densidade (HDL), diabetes melito e risco aumentado de doença vascular aterosclerótica.6,16
DMCPE1C. A DMCPE1C é causada por mutações no gene codificador da caveolina-3.19-23 A caveolina-3, uma proteína de 21 a 24 kDa da membrana interna, pode atuar na regulação da glicólise muscular. Os pacientes podem apresentar diferentes fenótipos clínicos: DMCM, hiper-CKmia isolada, doença do músculo ondulado ou miopatia distal.
DMCPE1D, 1E, 1F. Estes distúrbios representam mutações raras. A análise de linkage determinou os loci cromossômicos das mutações causais, porém os genes pertinentes não foram identificados.
Distrofias musculares do cíngulo dos membros autossômicas recessivas (DMCPED2)
DMCPE2A. A DMCPE2A é causada por mutações no gene codificador da calpaína-3.24 A deficiência de calpaína-3 é a forma mais frequente de DMCPE. A calpaína-3 é uma protease ativada por cálcio, que atua na diferenciação muscular.
A doença manifesta-se em pacientes com idade entre 8 e 30 anos. Os pacientes com deficiência de calpaína-3 apresentam enfraquecimento dos músculos do cíngulo dos membros inferiores, em especial dos glúteos, que poupa os abdutores do quadril. A formação de asas escapulares e o envolvimento da parte posterior da coxa são comuns. Os níveis séricos de CK geralmente estão elevados e às vezes ultrapassam 9.000 UI/L. As mutações no gene codificador da calpaína-3 foram identificadas e localizadas no cromossomo 15q15.1-q15.3.24 O diagnóstico é confirmado pelo ensaio de Western blot. Não existe nenhum tratamento específico para este distúrbio.
DMCPE2B. A DMCPE2B é causada por mutações no gene codificador da disferlina.25,26 A disferlina é uma proteína associada à membrana que não integra o complexo distrofina-glicoproteína [Figura 1]. A disferlina interage com a caveolina e pode estar envolvida no reparo da membrana. As mutações no gene da disferlina causam 2 tipos de miopatia: a miopatia de Miyoshi, que é caracterizada por níveis séricos de CK muito altos e envolvimento inicial do músculo gastrocnêmio; e a DMCPE2B, que pode se manifestar como um enfraquecimento muscular proximal e distal no final da adolescência ou início da 3ª década de vida. Mutações idênticas podem causar miopatia de Miyoshi ou DMCPE2B.25,26
Alguns pacientes com disferlinopatia apresentam um proeminente enfraquecimento distal, mas não exibem o fenótipo da miopatia de Miyoshi. As disferlinopatias são distúrbios comuns. Seu diagnóstico é confirmado pela detecção da ausência da disferlina no sarcolema de amostras de biópsia de músculo por imuno-histoquímica e com o uso de anticorpos antidisferlina. O defeito também pode ser observado em monócitos do sangue periférico no ensaio de Western blot. A ausência da disferlina pode resultar em perturbação da liberação da membrana e interferência no reparo de fibras musculares danificadas, possivelmente em consequência de defeitos no tráfico vesicular junto à fibra muscular.6,20,25,26 Não há tratamento específico para este distúrbio.
DMCPE2C, 2D, 2E, 2F. Estes 4 distúrbios são causados por mutações nos genes codificadores de 4 membros do complexo sarcoglicana: DMCPE2C (mutação no gene codificador da gamassarcoglicana, mapeada no cromossomo 5q33), DMCPE2D (alfassarcoglicana, mapeada em 17q12), DMCPE2E (betassarcoglicana, mapeada no cromossomo 4) e DMCPE2F (deltassarcoglicana, mapeada em 13q12).20,26,27 Nestes 4 distúrbios, os outros 3 componentes do complexo sarcoglicana são perdidos ou estão parcialmente ausentes, mas o complexo de distroglicana permanece normal.4,6,20,23,27
As mutações nos genes codificadores de sarcoglicanas incluem deleções missense, nonsense e in-frame, que resultam em um fenótipo de DMCPE de graus variáveis de severidade clínica. As mutações acarretam a montagem incorreta das sarcoglicanas com as proteínas do complexo de distroglicana, quebrando a ligação existente entre o sarcolema e a matriz extracelular [Figura 1].
Os pacientes apresentam enfraquecimento leve a severo da musculatura proximal (especialmente nas pernas), elevação dos níveis séricos de creatinina para cerca de 3.000 UI/L, alterações distróficas musculares e, com frequência, hipertrofia da panturrilha. A idade dos pacientes no momento do aparecimento da condição é variável. A análise de amostras de biópsia de músculo por imunocitoquímica e immunoblot demonstram a ausência ou severa deficiência de sarcoglicana. O grau de severidade do enfraquecimento muscular parece depender do grau de expressão de sarcoglicana residual. Há relatos de mutações nos genes correspondentes em até 60% dos pacientes. Também pode haver miocardiopatia, seja como manifestação isolada de miocardiopatia dilatada ou combinada à miopatia esquelética. Não há tratamento específico para este distúrbio.
DMCPE2G. A DMCPE2G é causada por mutações no gene codificador da teletonina, que é uma proteína do sarcômero localizada no disco Z do músculo esquelético, com consequente desorganização da estrutura sarcomérica.28 A DMCPE2G é um distúrbio da infância raro e relativamente brando, no qual não é raro haver envolvimento da musculatura distal.28 A biópsia de músculo pode mostrar a presença de vacúolos junto às fibras musculares. Os níveis séricos de CK são 10 a 30 vezes maiores do que os níveis normais. A ausência da teletonina, detectada por imunocitoquímica, confirma o diagnóstico.
DMCPE2H. A DMCPE2H é uma doença bastante rara, identificada apenas na população hutterite de Manitoba (Canadá). É causada por mutações no gene TRIM32 (o gene com motif tripartite), que está localizado no cromossomo 9q33.1.29,30
DMCPE2I. A DMCPE2I é causada por mutações no gene codificador da proteína relacionada à fukutina (FKRP), localizado no cromossomo 19q13.32.29,31 Este distúrbio e a distrofia muscular congênita de Fukuyama (DMCF) são distúrbios alélicos [ver Distrofia muscular congênita de Fukuyama (DMCF), adiante]. Ambos estão relacionados à ocorrência de alterações na expressão de alfadistroglicana (alfa-DG), que resulta de defeitos de glicosilação. A DMCPE2I é comum. Em alguns países, como no Reino Unido,31constitui uma das formas mais comuns de DMCPE.
O aparecimento da DMCPE2I é variável, podendo ocorrer no início da infância ou na fase adulta. Os pacientes apresentam enfraquecimento muscular proximal semelhante àquele observado na DMB, panturrilhas aumentadas, níveis séricos de CK elevados, língua aumentada (às vezes) e, muito frequentemente, envolvimento da musculatura respiratória e cardíaca. A inteligência permanece normal. A detecção de diminuição secundária da expressão de alfa-DG em uma biópsia de músculo ou a detecção de uma proteína de peso molecular reduzido no ensaio de immunoblot, aliada à diminuição dos níveis de alfa-2-lamina demonstrada por imunocitoquímica, conduzem à suspeita do diagnóstico de DMCM2I.
DMCPE2J. A DMCPE2J resulta de mutações no gene da titina, localizado no cromossomo 2q31. Este distúrbio, relatado na Finlândia,32 é alélico com a distrofia muscular tibial, que é uma doença autossômica dominante também relatada naquele país.
Distrofias musculares congênitas (DMC)
As DMC são distúrbios recessivos autossômicos caracterizados pelo desenvolvimento de enfraquecimento muscular no início da infância. Estas distrofias frequentemente estão associadas a malformações cerebrais e anormalidades cognitivas.33-35 Durante o período neonatal, os níveis séricos de CK são significativamente altos e muitas vezes chegam à ordem dos milhares. As DMC são distúrbios de migração neuronal referidos como córtex em “paralelepípedo” (cobblestone). Resultam na glicosilação anormal do complexo alfa-DG, com consequente rompimento da ligação entre a membrana e a matriz extracelular no músculo e no cérebro [Figura 1]. As DMC frequentemente são referidas como alfadistroglicanopatias [Tabela 1].
DMC1A. A DMC1A consiste na forma clássica de DMC e corresponde a mais de 40% dos casos. A doença está ligada ao cromossomo 6p22 e é causada por um defeito na merosina (alfa-2-lamina), que representa a espinha da estrutura da membrana basal[Figura 1]. Em contraste com outras formas de DMC, não há malformação cerebral nem retardo mental, embora este possa estar associado a uma leucoencefalapatia definida por imagem de ressonância magnética (IRM) (sem sinais evidentes de deterioração intelectual). A doença ocorre em crianças, mas também acomete adultos jovens. Os pacientes apresentam uma deficiência de alfa-2-lamina lâmina basal do músculo esquelético.4,6,35 Alguns pacientes apresentam uma deficiência parcial de merosina, resultante de mutação branda ou causas secundárias que são mais frequentemente mutações nos genes codificadores de fukutina ou FKRP.
Os pacientes apresentam hipotonia neonatal, níveis séricos de CK elevados, referenciais motores retardados, neuropatia axonal e enfraquecimento muscular respiratório. O grau de fenótipo clínico varia de moderado a severo. Em pacientes deficientes de alfa-2-lamina, o complexo distroglicana está desorganizado. Em consequência, a membrana muscular torna-se defeituosa.4,6,35 Como a alfa-2-lamina, a beta-1-lamina e a gama-1-lamina também estão presentes nas células de Schwann, alguns pacientes com DMC apresentam achados neuropáticos.
DMC1B. A DMC1B é causada por mutações no gene da alfa-7-integrina, localizado no cromossomo 12q13, que resultam na ausência de alfa-7-integrina a partir do sarcolema.36 A manifestação clínica é idêntica à manifestação clínica da DMC1A.
DMC1C. A DMC1C é uma doença rara, causada por mutações no gene FKRP, localizado no cromossomo 19q13.3. A DMC1C e a DMCM2I são distúrbios alélicos. O aparecimento do enfraquecimento muscular ocorre na 1ª semana após o nascimento. As crianças afetadas não conseguem andar de maneira independente. A doença é marcada pelo envolvimento respiratório, e a inteligência pode permanecer normal. A suspeita desta condição é levantada pela observação de uma diminuição variável ou ausência de alfa-DG no músculo, ou diante da detecção de uma alfa-DG com tamanho reduzido no ensaio de immunoblot.33,34
DMC1D. A DMC1D é produzida por uma mutação no gene LARGE. Trata-se de um distúrbio raro que resulta em retardo mental severo e distrofia. O diagnóstico é confirmado pela ausência de alfa-DG na biópsia de músculo.33,34
Distrofia muscular congênita de Fukuyama (DMCF). Este distúrbio representa a DMC mais comum entre os descendentes de japoneses. A DMCF é causada por uma mutação no gene da fukutina, com consequente deficiência desta proteína e diminuição da glicosilação de alfa-DG que é indicada por uma coloração mais fraca da alfa-DG no músculo. Os pacientes apresentam malformações cerebrais, retardo mental profundo e anormalidades oftalmológicas. A doença surge antes do 9 meses de idade, e os pacientes jamais aprendem a andar. Os indivíduos acometidos por DMCF em geral morrem ao redor dos 20 anos.33,34
Doença do músculo-olho-cérebro (MOC). A doença do músculo-olho-cérebro (MOC) é causada pela mutação no gene POMGnT da glicosiltransferase, que resulta em uma deficiência de alfa-DG confirmada por imunocitoquímica. Os pacientes apresentam hipotonia, enfraquecimento, hidrocefalia leve a moderada, hipoplasia cortical ou cerebelar e anormalidades no olho (p. ex., miopatia, microftalmia e hipoplasia do nervo óptico).33-36
Síndrome de Walker-Warburg. A síndrome de Walker-Warburg resulta de mutações no gene POMGnT1 da O-manosil transferase. Esta é a mais severa das DMC, compartilhando características clínicas com a DMCF e a MOC. Os pacientes geralmente morrem ao redor dos 3 anos de idade.33-36
DMC com contraturas articulares. Existem 3 formas de DMC caracterizadas pelo aparecimento de contraturas articulares. Os pacientes que apresentam estes distúrbios não apresentam defeito de glicosilação nem retardo mental. Estes distúrbios são DMC acompanhadas de síndrome da espinha rígida (DMER), miopatia de Ullrich e miopatia de Betlem. Não existe nenhuma terapia específica disponível.
A DMER resulta de mutações no gene codificador da selenoproteína N (SEPN1) no cromossomo 1q35-36.37-39 Os pacientes apresentam enfraquecimento muscular, hipotonia, enfraquecimento muscular e uma rigidez espinal que resulta em incapacidade de flexionar o pescoço, escoliose e dificuldades respiratórias.
A miopatia de Ulrich é causada por mutações que afetam uma das cadeias polipeptídicas formadoras de colágeno VI, necessário à interação com a matriz extracelular.38 Estes pacientes apresentam mobilidade distal excessiva combinada a contraturas proximais, espinha rígida ao nascimento e enfraquecimento muscular. A biópsia de músculo revela a ausência de colágeno VI. As miopatias de Ulrich e Betlem são distúrbios alélicos.
A miopatia de Betlem, uma doença autossômica dominante, é causada por mutações que afetam subunidades de proteínas da matriz extracelular (colágeno VI-alfa-1, VI-alfa-2 e VI-alfa-3).37 O aparecimento da doença ocorre na infância ou na adolescência. Entre as manifestações clínicas estão o enfraquecimento muscular leve e as contraturas em múltiplas articulações, que podem estar presentes ao nascimento.
Miopatias miofibrilares (MMF) decorrentes de mutações em proteínas do filamento intermediário
Os filamentos intermediários exercem papel decisivo na promoção da integração mecânica das miofibrilas e na proteção das fibras musculares contra o estresse mecânico repetitivo. As MMF constituem um grupo recém-identificado de distúrbios geneticamente heterogêneos, que afetam os filamentos intermediários da musculatura. Os pacientes compartilham os aspectos miopatológicos da desintegração das miofibrilas, bem como o acúmulo de produtos de degradação no interior de inclusões que contêm desmina e outras proteínas miofibrilares e ectópicas.40-44 Um aspecto forte que une todos estes distúrbios etiologicamente diversos reside no fato de a desintegração miofibrilar começar ou envolver subsequentemente o disco Z do sarcômero. As proteínas envolvidas na causa da MMF variam significativamente quanto à estrutura e função. Entre estas proteínas, estão[Figura 1]:
1. Desmina – proteína de filamento intermediário de tipo III. Interliga as miofibrilas ao nível do disco Z e conecta-as a outras organelas celulares, ancorando o citoesqueleto. No coração, a desmaninha está presente em maior concentração nos discos intercalados e constitui o principal componente das fibras de Purkinje.
2. Alfa-B-cristalina – membro da família de proteínas do choque térmico. Atua como chaperona para a desmina.
3. Miotilina – proteína do disco Z do sarcômero, que apresenta expressão forte no músculo esquelético e expressão fraca no miocárdio. Exerce papel significativo na montagem do sarcômero.
4. ZASP – um dos principais componentes do disco Z, que mantém sua integridade estrutural durante a contração.
5. Filamina C – apresenta reação cruzada com a actina, ao nível do disco Z, e liga-se a outras proteínas do disco ao interagir com o complexo distrofina-distroglicana no sarcolema.
6. BAG3 – participa das vias antiapoptóticas e é fortemente expressa nos músculos esquelético e cardíaco. As mutações ocorridas nos genes codificadores das proteínas do citoesqueleto que sustentam o disco Z e o sarcômero foram implicadas como causa de doenças com denominações correspondentes: clesminopatia, alfa-B-cristalinopatia, miotilinopatia,21ZASPopatia, C-filaminopatia e BAG3opatia.40-44
Em geral, as MMF constituem um grupo importante e muitas vezes negligenciado de distúrbios que se manifestam como defeitos de condução ou como uma miopatia esquelética de aparecimento distal e progressão para os músculos proximais, faciais ou respiratórios.43 A suspeita diagnóstica é considerada diante da observação de acúmulo de produtos miofibrilares na biópsia de músculo, evidenciado por colorações histoquímicas enzimáticas. O diagnóstico, então, é confirmado por análise de mutação.
Não existe tratamento específico para estes distúrbios, contudo o reconhecimento precoce pode permitir a identificação de potenciais candidatos ao recebimento de um marca-passo, com a finalidade de prevenir mortes súbitas em consequência de arritmias.
Distrofias autossômicas dominantes com fenótipo único
Distrofia miotônica
A distrofia miotônica é a distrofia muscular mais comum em pacientes adultos. Sua incidência é de 1 em cada 8.000 indivíduos, e a prevalência aproximada é de 5 em cada 100.000 indivíduos. O distúrbio apresenta 2 subclassificações: distrofia miotônica 1 (é a forma clássica de distrofia miotônica, doença de Steinert) e distrofia miotônica 2 (miopatia miotônica proximal [MMPRO]) [Tabela 1]. Distrofia miotônica 1 e 2 são síndromes de múltiplos órgãos autossômicas dominantes. Ambas apresentam similaridades marcantes em termos de manifestações clínicas.45,46
A distrofia miotônica apresenta uma distribuição exclusiva: (1) ptose das pálpebras, sem envolvimento da musculatura extraocular; (2) atrofia dos músculos masseter e temporal, que confere uma configuração facial estreitada única; (3) atrofia do músculo esternocleidomastoideo, com uma relativa preservação dos músculos da cintura escapular e cervical posterior (um sinal clínico que diferencia a distrofia miotônica da distrofia muscular fascioescapuloumeral [DFEU]); (4) atrofia do grupo de músculos distais, com discreto envolvimento proximal durante os estágios iniciais da doença; e (5) envolvimento dos músculos palatal e faríngeo, que pode acarretar disartria e disfagia.4,6,45,46
A miotonia, definida pelo retardo do relaxamento de uma contração muscular normal, é importante como sinal clínico. Para deflagrar a miotonia durante o exame, solicita-se ao paciente que realize um aperto de mão forte e solte rapidamente, em seguida. Na miotonia, é evidente a incapacidade de liberar o aperto de mão imediatamente. A percussão da eminência tenar ou do extensor dos dedos também revela o relaxamento lento característico da miotonia. Entre os aspectos sistêmicos estão os defeitos de condução cardíaca, disfunção mental leve (muitas vezes, com expressões e comportamentos inconvenientes ou estúpidos), atrofia testicular, calvície frontal, catarata, envolvimento do trato gastrintestinal (com retardo da motilidade e do esvaziamento), hiperinsonia e resposta diminuída à hipóxia, acarretando falta de concentração e apatia. Do ponto de vista clínico, a distrofia miotônica 2 é semelhante à distrofia miotônica 1 do adulto. Entretanto, o grau de expressão das características sistêmicas pode ser variável em relação ao observado na distrofia miotônica 1. Os aspectos mais importantes na distinção entre distrofia miotônica 1 e distrofia miotônica 2 são: o envolvimento muscular proximal preferencial na distrofia miotônica 2; a falta de formas congênitas de distrofia miotônica 2; a incidência rara de antecipação (ver adiante) na distrofia miotônica 2; e a incidência rara de disfunção cognitiva na distrofia miotônica 2.
As crianças de mães afetadas podem apresentar diminuição dos movimentos fetais e sintomas logo nas primeiras fases da vida, tais como hipotonia severa, dificuldade para se alimentar, enfraquecimento facial bilateral e angústia respiratória (distrofia miotônica congênita). A distrofia miotônica deve ser distinguida da miotonia congênita, as quais seguem padrões de herança recessiva e dominante. Os pacientes com miotonia congênita apresentam miotonia e, com frequência, hipertrofia muscular. Em contraste com a distrofia miotônica, contudo, a miotonia congênita não está associada ao enfraquecimento muscular, atrofia nem sintomas sistêmicos. Trata-se de uma condição resultante de um defeito genético diferente que afeta os canais de cloreto [ver Patologias de canais iônicos, paralisias periódicas e miotonias não distróficas, adiante].
O diagnóstico clínico de distrofia miotônica é confirmado por eletromiografia (EMG), que mostra as descargas miotônicas. Em casos difíceis, os exames com lâmpada de fenda podem mostrar a formação de catarata ainda no início. A expressão clínica da distrofia miotônica é variável, e o distúrbio pode permanecer não diagnosticado até os pacientes terem filhos. Na distrofia miotônica 1, a idade do paciente no momento do aparecimento da doença é progressivamente menor em gerações sucessivas (um fenômeno conhecido como antecipação). É comum encontrar famílias nas quais o único sintoma apresentado pela avó foi uma catarata em estágio inicial, embora sua filha (que agora sabe estar afetada pela condição) tenha dado à luz um bebê com distrofia miotônica congênita severa. A antecipação é comum na distrofia miotônica 1 e é rara na distrofia miotônica 2.
A distrofia miotônica 1 é causada pela presença de uma sequência repetitiva do trinucleotídeo CTG (citosina, timina, guanina) na região codificadora não proteica do gene de uma proteína quinase denominado DMPK, localizado no cromossomo 19.45-48 Em indivíduos levemente afetados, a região de repetição CTG polimórfica presente no gene de proteína quinase sofre uma expansão de 50 a 80 repetições. Em indivíduos severamente afetados, pode haver mais de 2.000 repetições. O tamanho da expansão CTG aumenta ao longo das gerações e isto contribui para a antecipação. As medidas do comprimento da expansão CTG podem ser usadas para confirmar a presença de distrofia miotônica em familiares de um paciente, estabelecer um diagnóstico pré-natal ou no aconselhamento genético efetivo de indivíduos assintomáticos com risco de distrofia miotônica.
A distrofia miotônica 2 também é causada por uma repetição expandida em uma região codificadora não proteica envolvendo uma repetição CTG no gene ZNF9 (proteína em dedo de zinco 9), no cromossomo 3q21, que codifica um fator de transcrição. O fator patogênico da distrofia miotônica 1 e da distrofia miotônica 2 é o RNA produzido a partir do gene mutante, e não a proteína. Esta é a 1ª doença comprovadamente causada por um RNA prejudicial.45 O RNA mutante forma inclusões no núcleo (inclusões ribonucleares). Foi demonstrado que a repetição existente no RNA se liga a proteínas da família muscleblind e interfere na função normal de processamento do RNA.48 Estes dados sugerem que a terapia para distrofia miotônica deve estar voltada para a eliminação do RNA prejudicial.
Atualmente, a terapia para distrofia miotônica é sintomática. O suporte emocional e a educação sobre as precauções necessárias para evitar quedas e lesões são essenciais. O monitoramento atento da condição cardíaca, especialmente durante a administração da anestesia, é importante. Fármacos como a quinidina, procainamida, mexiletina, fenitoína e os betabloqueadores podem ajudar a aliviar a miotonia, mas não amenizam o enfraquecimento. Dentre estes agentes, a mexiletina parece ser o mais efetivo. A testosterona falhou como terapia para distrofia miotônica. O mono-hidrato de creatina pode proporcionar alívio mínimo da mialgia. O modafinil pode diminuir a sonolência excessiva durante o dia, melhorar o humor e diminuir a fadiga.
Distrofia muscular fascioescapuloumeral (DFEU)
A DFEU é a 3ª forma mais comum de distrofia muscular. Seu início geralmente ocorre durante a 2ª década da vida.
A DFEU está ligada ao cromossomo 4q. Trata-se de uma doença autossômica dominante, porém 25% dos casos resultam de mutações novas.49,50 Esta doença apresenta uma heterogeneidade significativa. Alguns pacientes não apresentam os aspectos típicos da DFEU (ver adiante) e, em vez disso, podem exibir miopatias distais características de DMCPE. Entre os familiares de um paciente, há alguns em que o envolvimento da musculatura facial pode ser apenas mínimo. O mosaicismo de linhagem germinativa ocorre em 10% das famílias. Isto significa que mais de um irmão é afetado em uma determinada geração, na ausência de envolvimento de ambos os pais.49,50
Os pacientes apresentam enfraquecimento da musculatura facial (especialmente do músculo orbicular do olho), enquanto os músculos extraoculares e masseter são poupados. O enfraquecimento inicial dos músculos escapulares resulta na formação de asas escapulares proeminentes e confere aos ombros uma aparência inclinada para a frente. O enfraquecimento dos músculos tibiais anteriores, que resulta no pé caído, está sempre presente. A doença evolui lentamente, e há períodos longos de estabilidade. A atrofia da língua é um fenômeno comum. A progressão ocorre de forma descendente: o envolvimento dos músculos da cintura escapular é seguido do envolvimento do bíceps, tríceps e músculos da cintura pélvica. A ampla maioria dos pacientes apresenta anormalidades capilares, descolamento da retina e comprometimento da audição. Estes achados são mais frequentes na forma infantil da DFEU.
Na DFEU, os níveis séricos de CK estão levemente aumentados, e o miocárdio é poupado. Os achados do exame de biópsia de músculo são variáveis e podem incluir a presença de células inflamatórias. A suspeita diagnóstica baseia-se na clínica e é confirmada pela análise do DNA. A doença é causada pela ocorrência de uma deleção junto a uma série de repetições de 3,3 kb (D4Z4), no cromossomo 4. Quando o número de repetições se torna inferior a um número de repetições crítico (aproximadamente 10), ocorre a expressão clínica do gene. Cerca de 95% dos pacientes apresentam uma deleção que resulta em um pequeno fragmento de DNA, com comprimento menor que 35 kb, detectado com auxílio de algumas enzimas de restrição.49
Em estudos controlados, o uso de prednisona ou albuterol (um agonista beta-2-adrenérgico) não proporcionou benefícios no tratamento da DFEU, embora este último possa aumentar a massa muscular.50
Distrofia muscular oculofaríngea (DMOF)
A distrofia muscular oculofaríngea (DMOF) é uma doença autossômica dominante rara, que se manifesta entre a 4ª e a 6ª décadas da vida. É caracterizada por ptose e disfagia, que podem ser ambas severas. Pode haver um leve enfraquecimento da musculatura distal. A mutação responsável pela DMOF é causada pela repetição de expansão do trinucleotídeo GCG no 1º éxon do gene codificador da proteína ligadora de poli(A)-2 (PABP2), junto ao cromossomo 14q11,2. O gene PABP2 está localizado em agregados intramusculares de fibras musculares e possui uma relação causal com estas inclusões.51
Hipertermia maligna (HM)
A hipertermia maligna (HM) ocorre em 1 a cada 50.000 a 100.000 indivíduos adultos durante a anestesia geral, sobretudo com o uso de halotano isolado ou em combinação com succinilcolina e outros relaxantes musculares despolarizantes. É caracterizada por um rápido aumento do metabolismo aeróbico e anaeróbico, quando a temperatura corporal pode ultrapassar 43°C. A HM manifesta-se com taquicardia, rigidez muscular (causada por uma contratura muscular que pode evoluir para rigidez ou morte), aumento da permeabilidade muscular (resultando em níveis séricos aumentados de potássio [K+], cálcio [Ca2+] e sódio [Na+], além de edema), liberação excessiva de mioglobulina a partir do músculo e mioglobinúria. O trismo ou espasmo do músculo masseter, que ocorre durante a indução de anestesia, pode ser indicativo de HM.
Embora a HM ocorra em alguns pacientes sem doença muscular comprovada, os pacientes considerados de risco são aqueles com múltiplas anormalidades musculoesqueléticas congênitas, deslocamento de quadril congênito isolado ou doença envolvendo o núcleo central. Também foi comprovado que a HM ocorre em alguns pacientes com DMD ou DMB. A suscetibilidade à HM é hereditária como traço autossômico dominante e parece ser precipitada pela incapacidade de controlar as concentrações de cálcio junto às fibras musculares, em decorrência do mau funcionamento do retículo sarcoplasmático (RS). Em até 50% dos heredogramas, os alelos mutantes são encontrados no receptor de rionidina (RyR),52 que une o hiato existente entre o RS e o túbulo transverso, ou no gene CACNA1S, que codifica a subunidade alfa-1 do canal de cálcio voltagem-dependente do tipo L di-hidropiridina-sensível.52
A HM deve ser distinguida da rabdomiólise pós-anestésica subsequente ao estresse muscular, reação tóxica a fármacos, tempestade tireóidea precipitada por cirurgia e anestesia e, ainda, síndrome maligna neuroléptica precipitada por fármacos psicoativos (p. ex., haloperidol e fenotiazinas), que bloqueiam as vias dopaminérgicas centrais.52,53
O dantrolene é efetivo como tratamento para HM. Este fármaco diminui a liberação de cálcio a partir do RS, sem alterar sua recaptação. O episódio agudo é tratado de maneira sintomática. O dandrolene endovenoso (2 a 10 mg/kg, a cada 5 minutos) deve ser administrado no início do episódio, enquanto a perfusão muscular ainda é adequada. Para os pacientes comprovadamente suscetíveis à HM, o dandrolene pode ser administrado a uma dosagem de 2 mg/kg, em 10 a 15 minutos antes da administração de anestesia. A melhor forma de prevenir os episódios de HM em indivíduos suscetíveis é o uso de anestésicos seguros (p. ex., óxido nitroso e tiopental) e relaxantes musculares não despolarizantes.
Miopatias metabólicas
Princípios de energia muscular
Como um grupo, as miopatias metabólicas são caracterizadas pela deficiência de produção de energia causada por distúrbios envolvendo glicogênio, lipídio ou mitocôndria.
As 2 fontes principais de energia para o músculo são o glicogênio e os ácidos graxos, cujas vias metabólicas convergem na via da acetil coenzima A (acetil-CoA), para oxidação final junto à mitocôndria, por meio do ciclo de Krebs e da cadeia respiratória.54-56 De resto, a energia muscular deriva principalmente da oxidação de ácidos graxos livres (AGL). Durante o exercício aeróbico de alta intensidade, o glicogênio constitui a principal fonte de combustível para a oxidação fosforilativa. As reservas de glicogênio musculares são depletadas após 90 minutos de exercício. Com o exercício prolongado, o uso dos AGL e da glicose sanguínea é intensificado. Como a disponibilidade de AGL provenientes do tecido adiposo é quase ilimitada, um indivíduo saudável consegue realizar exercícios de intensidade moderada durante várias horas. Em pacientes com miopatias metabólicas, os sintomas tornam-se evidentes durante as atividades que envolvem demandas metabólicas aumentadas, como o exercício físico. As miopatias metabólicas resultam de defeitos do metabolismo e uso do glicogênio, glicogênio ou lipídio pelo músculo. As miopatias resultantes do uso de glicogênio e glicose são classificadas de acordo com a sequência de defeitos enzimáticos ao longo das vias glicogenolíticas ou glicolíticas [Figura 2] e caracterizados pela intolerância ao exercício, mialgia, cãibras e, por fim, mioglobinúria. Alguns pacientes podem desenvolver um enfraquecimento progressivo e fixo. As 2 categorias principais de miopatias metabólicas são aquelas devidas à doença de armazenamento de lipídio ou glicogenose.
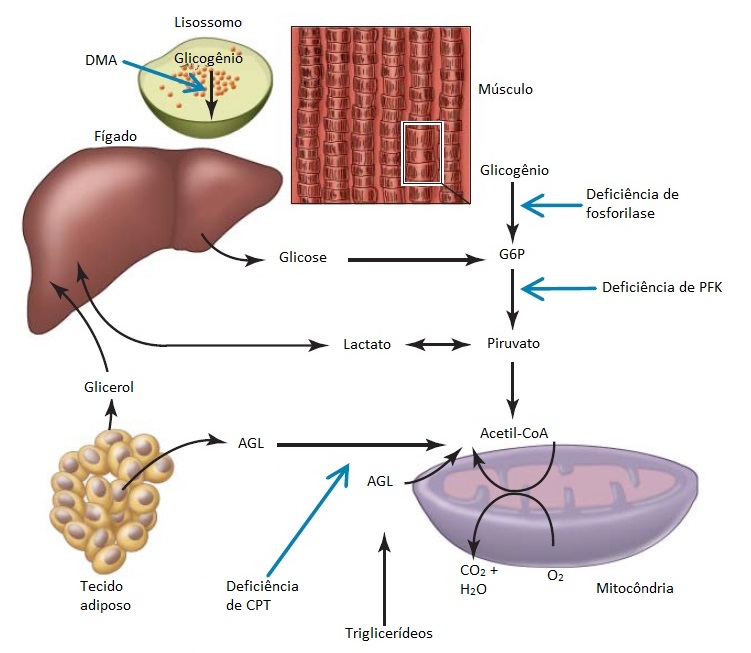
Figura 2. Esquema representativo do metabolismo do glicogênio, glicólise e uso de ácidos graxos. Algumas das miopatias mais comuns são aquelas resultantes de deficiências das enzimas maltase ácida, fosforilase muscular, fosfofrutoquinase (PFK) e carnitina palmitoiltransferase (CPT).
AGL = ácidos graxos livres; DMA = deficiência de maltase ácida.
Glicogenoses
Deficiência de fosforilase muscular
A deficiência de fosforilase muscular (também conhecida como doença de McArdle) é o protótipo da glicogenose: a quebra do glicogênio é inibida e isto resulta na falta de piruvato e comprometimento do débito energético [Figura 2]. Trata-se da 2ª causa mais comum de mioglobinúria recorrente, após a deficiência de carnitina palmitoiltransferase (CPT).54-56
Esta deficiência é uma doença autossômica recessiva, que se manifesta em pacientes com mais de 15 anos de idade sob a forma de intolerância ao exercício e mioglobinúria. Quando o paciente descansa brevemente ao apresentar rigidez e mialgia induzidas pelo exercício, consegue retomar a atividade com maior resistência (fenômeno do 2º fôlego) devido ao aumento da mobilização e do uso dos AGL e da glicose. O enfraquecimento muscular fixo pode se desenvolver em fases mais tardias da vida. Os níveis séricos de CK de repouso frequentemente estão elevados. A incapacidade de produzir lactato venoso após o exercício é tradicionalmente avaliada pelo teste do exercício do antebraço isquêmico. Este teste, contudo, está caindo em desuso como ferramenta diagnóstica, porque fornece resultados falso-positivos, é inespecífico, pode ser doloroso e pode resultar em dano muscular focal. Um teste de exercício não isquêmico tem o mesmo valor diagnóstico e está livre das desvantagens associadas ao teste isquêmico. A biópsia de músculo mostra a ausência de fosforilase, a presença de vacúolos subsarcolêmicos e o acúmulo aumentado de glicogênio. O diagnóstico é confirmado pela análise bioquímica do músculo e pela análise molecular das células sanguíneas. O defeito é causado por mutações que afetam a isoforma muscular da fosforilase, junto ao cromossomo 11q13 e pode ser detectado nos leucócitos em mais de 90% dos pacientes.
Não existe tratamento disponível para a doença de McArdle, porém o treinamento com exercícios aeróbicos aliado a uma dieta rica em proteínas pode ser útil. A carga de sucrose antes do exercício pode melhorar a tolerância ao exercício e conferir proteção contra a rabdomiólise exercício-induzida.57
Deficiência de fosfofrutoquinase (PFK)
A deficiência de fosfofrutoquinase (PFK) é uma doença autossômica recessiva causada por mutações distintas que afetam a subunidade M, localizadas no cromossomo 1. Trata-se de um defeito glicolítico [Figura 2] com consequências funcionais similares àquelas observadas na doença de McArdle. A suspeita de deficiência de PFK deve ser considerada em casos de pacientes que apresentam intolerância ao exercício, náusea e mioglobinúria. Uma história longa de hemólise branda e compensada, contagem de reticulócitos alta, níveis de bilirrubina elevados e hiperuricemia também apontam a ocorrência de deficiência de PFK, especialmente em alguns grupos étnicos, como os japoneses e judeus de Ashkenazi. O diagnóstico é confirmado por exames bioquímicos do músculo e pela análise molecular das células sanguíneas.58 Não há tratamento específico. Os pacientes devem evitar refeições com alto teor de carboidrato. A adoção de uma dieta cetogênica tem sido defendida.
Deficiência de fosfoglicerato quinase (PGK)
A deficiência de fosfoglicerato quinase (PGK) é um raro distúrbio recessivo ligado ao X, que se manifesta como intolerância ao exercício, episódios de mioglobinúria, anemia hemolítica, miopatia e, ocasionalmente, retardo mental leve54-56 [Figura 2].
Deficiência de fosfoglicerato mutase
A deficiência de fosfoglicerato mutase (PGAM) é uma doença bastante rara, que afeta apenas a musculatura. Nos Estados Unidos, foi identificada somente em afro-americanos [Figura 2].
Deficiência de maltase ácida (DMA)
A deficiência de maltase ácida (DMA) é uma doença autossômica recessiva de armazenamento de glicogênio. É causada pela deficiência de alfaglicosidase (GAA), uma enzima que degrada o glicogênio lisossômico e é codificada por um gene localizado no cromossomo 17q23.54-57,59 As mutações ou pequenas deleções causadoras de splicing anormal afetam a expressão de GAA.59,60
Existem 3 formas clínicas de DMA: do 1º ano de vida, da infância e do adulto.59,60 A forma associada ao 1º ano de vida (doença de Pompe) surge durante os primeiros meses de vida e manifesta-se como hipotonia, enfraquecimento e ampliação do coração, língua e fígado. As alterações respiratórias e cardiovasculares levam à morte do paciente antes que complete 2 anos de idade.
Na forma infantil da DMA, os pacientes apresentam miopatia caracterizada pelo retardo dos referenciais motores, enfraquecimento da musculatura proximal, envolvimento da musculatura respiratória e aumento da panturrilha. A doença leva à morte do paciente ao redor da 2ª década da vida.
A forma adulta da DMA manifesta-se em indivíduos com mais de 20 anos de idade, sob a forma de um enfraquecimento da musculatura proximal semelhante à polimiosite ou à distrofia do cíngulo dos membros. O enfraquecimento dos músculos respiratórios pode ser o sintoma observado em 1/3 dos pacientes adultos com DMA. O acúmulo de glicogênio ocorre de forma predominante no músculo. Entretanto, a deficiência enzimática ocorre no músculo, fígado, coração e em fibroblastos mantidos em cultura. Os pacientes apresentam níveis séricos elevados de CK. A EMG mostra descargas miotônicas proeminentes (sem miotonia clínica), em especial nos músculos paraespinais. A biópsia de músculo mostra a presença de múltiplos vacúolos contendo altas concentrações de glicogênio que reagem fortemente com a fosfatase ácida, indicando uma atividade lisossômica aumentada. Como a utilização do glicogênio e da glicose não é comprometido[Figura 2], a DMA promove um enfraquecimento fixo sem intolerância ao exercício nem mioglobinúria. A fibra muscular passa por um processo autofágico devido às anormalidades lisossômicas.
O diagnóstico é confirmado por análise genética molecular, e é importante que este teste seja realizado prontamente, a fim de possibilitar a instituição do tratamento com terapia de reposição enzimática. Em alguns estudos, incluindo um estudo controlado recente,60 foi demonstrado que o tratamento com GAA estava associado à melhora da distância caminhada e à estabilização da função pulmonar ao longo de um período de 18 meses.
Outras glicogenoses raras
As glicogenoses musculares raras incluem os distúrbios causados por deficiências de enzimas musculares específicas. Entre estes distúrbios estão as deficiências de lactato desidrogenase (LDH), betaenolase, enzima desramificadora, enzima ramificadora e aldolase.54-56
Miopatias de armazenamento lipídico
Durante o exercício contínuo, os ácidos graxos de cadeia longa (AGCL) constituem a principal fonte de energia para o músculo. Os AGCL derivam dos alimentos ou, sob condições de jejum, do tecido adiposo. Os AGCL primeiro devem ser transportados até a mitocôndria para serem oxidados. Sua transferência através da membrana mitocondrial interna requer L-carnitina e 2 enzimas – CPT I e II – que estão localizadas junto às membranas mitocondriais externa e interna, respectivamente. Dentro da mitocôndria, a alfaoxidação é facilitada principalmente pelas acil coenzima A (acil-CoA) desidrogenases e, em seguida, pela transferência de elétrons via flavoproteínas para as proteínas da cadeia respiratória.54-56 As miopatias de armazenamento lipídico são causadas pelo comprometimento da oxidação dos ácidos graxos pela mitocôndria, que resulta de defeitos envolvendo (1) a carnitina e a CPT, comprometendo o transporte de ácidos graxos através da membrana mitocondrial; (2) as enzimas associadas à betaoxidação; e (3) as proteínas da cadeia respiratória e flavoproteínas transferidoras de elétrons.54
Deficiência de carnitina
A carnitina é mais frequentemente derivada da dieta, porém 25% de toda a carnitina é sintetizada no fígado, a partir de lisina e metionina. A carnitina é essencial à oxidação dos AGCL.54 O ônus gerado pela deficiência de carnitina é a disfunção dos rins, coração e tecidos musculares, que são altamente dependentes da oxidação de AGCL.
A deficiência de carnitina primária (DCP) é um distúrbio autossômico recessivo incomum que ocorre durante a infância e é causado por mutações no gene SLC22A5, codificador do transportador de cátions dependente de íon sódio-2 (OCTN2). Estas mutações causam deficiência do número de receptores de carnitina de alta afinidade funcionais, resultando no aparecimento de defeitos de transporte da carnitina através das membranas celulares.54 As causas mais comuns de deficiência de carnitina são secundárias e resultam de: (1) alfaoxidação defeituosa associada a acidúrias orgânicas; (2) disfunção mitocondrial; (3) doença renal (p. ex., síndrome de Fanconi, cistinose nefropática ou hemodiálise crônica); e (4) tratamento farmacológico, especialmente com zidovudina (AZT) e valproato.61 Os pacientes com DCP apresentam miocardiopatia progressiva, episódios de hipoglicemia hipocetótica (decorrentes da disfunção hepática) e enfraquecimento miopático proximal. Os lipídios acumulam-se na musculatura e formam pequenas gotículas de gordura. O tratamento com suplementação de carnitina produz resultados variáveis.
Deficiência de carnitina palmitoiltransferase (CPT)
Em bebês, a deficiência de CPT I manifesta-se como síndrome de Reye, com encefalopatia hepática, hipoglicemia hipocetótica e hiperamonemia. Em adultos, a síndrome de deficiência de carnitina resulta mais frequentemente da deficiência de CPT II, causada por mutações no gene da CPT II, que está localizado no cromossomo 11p11-p13.54-56 A deficiência de CPT constitui a causa mais comum de mioglobinúria em adultos jovens. Os pacientes apresentam ataques de rigidez muscular, cãibras, mialgia e mioglobinúria decorridas algumas horas da prática prolongada ou contínua de exercícios, sobretudo após o jejum ou quando o fornecimento de energia para os músculos depende do uso dos AGCL e não da utilização de glicogênio ou glicose. Os pacientes deficientes de CPT II não apresentam diminuição da tolerância ao exercício, fenômeno do 2º fôlego nem os sinais de alerta de mialgia que impedem a continuidade da prática de exercício. Entre os ataques, a força muscular e os níveis séricos de CK permanecem normais. O diagnóstico é estabelecido pela determinação da atividade de CPT II no músculo ou por meio do teste genético. Na maioria dos casos, a biópsia de músculo não é reveladora.
Não se sabe por que a deficiência de CPT causa ataques intermitentes de mioglobinúria. Também é desconhecido o motivo pelo qual não há acúmulo tardio de lipídios na musculatura.54-56 Não existe terapia para prevenção dos ataques de mioglobinúria. Uma dieta com alto teor de carboidratos e pobre em gorduras, refeições frequentes e ingesta de carboidrato extra antes e durante a prática contínua de exercícios são as ações recomendadas.
Encefalopatias e miopatias mitocondriais
As encefalopatias e miopatias mitocondriais constituem um grupo diverso de distúrbios que afetam não só os músculos e o sistema nervoso, mas também outros órgãos. Estes distúrbios são caracterizados por um defeito primário envolvendo o débito energético. Os defeitos genéticos de enzimas energéticas mitocondriais podem ser causados por mutações no DNA mitocondrial (mtDNA) ou no DNA nuclear. Existem 3 tipos de mutações responsáveis por este grupo de doenças variadas:62-65 (1) mutações esporádicas no mtDNA, que causam deleções em ampla escala no mtDNA e são responsáveis por distúrbios multissistêmicos (p. ex., síndrome de Kearns-Sayre [SKS]) com possíveis efeitos sobre o coração, cérebro, sistema endócrino e trato gastrintestinal; (2) mutações pontuais no mtDNA herdado da mãe, que afetam o cérebro e a musculatura causando distúrbios como a síndrome MELAS (encefalopatia mitocondrial, acidose láctica e episódios do tipo acidente vascular cerebral), síndrome MERRF (epilepsia mioclônica e miopatia com fibras vermelhas esfarrapadas) e neuropatia óptica hereditária de Leber (NOHL); e (3) depleção tecido-específica do mtDNA, que leva ao desenvolvimento de uma síndrome multi-órgãos com possíveis efeitos sobre os músculos, fígado, rins e cérebro.
A principal função da mitocôndria consiste em gerar energia para a célula. Para tanto, a mitocôndria produz trifosfato de adenosina (ATP) por fosforilação oxidativa (FOX), que envolve 5 complexos enzimáticos (designados por I, II, III, IV [citocromo-c oxidase] e V) localizados na membrana mitocondrial interna.62-65 As mitocôndrias contêm seu próprio DNA extracromossômico (mtDNA), que difere do DNA nuclear. A organização do mtDNA é altamente compacta, e não há íntrons. Em consequência, as mutações que ocorrem ao acaso no mtDNA geralmente atingem uma sequência codificadora e com frequência causam doença. Além disso, o mtDNA é suscetível ao dano produzido pelos radicais do oxigênio, por estar nas proximidades do local de produção destas moléculas por FOX e apresentar mecanismos mínimos de reparo.62-65 O mtDNA inteiro de cada indivíduo é herdado exclusivamente da mãe (herança não mendeliana), pois o espermatozoide contribui apenas com DNA nuclear para o zigoto, durante a fecundação. A FOX também pode sofrer diminuição como resultado da ocorrência de mutações nos genes de FOX codificados no núcleo. Neste caso, as doenças seguem um padrão de herança mendeliana.
O exame da biópsia de músculo de pacientes com defeitos de FOX resulta anormal e revela a presença de fibras vermelhas esfarrapadas coradas com tricrômio ou fibras azuis esfarrapadas coradas por succinato desidrogenase. Estes achados resultam do acúmulo de mitocôndrias na periferia das fibras musculares, bem como do acúmulo de fibras negativas para citocromo-c oxidase. À microscopia eletrônica, as mitocôndrias apresentam inclusões paracristalinas ou cristas anormais. As mutações, deleções ou depleções específicas são detectadas pelo exame do mtDNA.
Deleções esporádicas de DNA mitocondrial (mtDNA)
Síndrome de Kearns-Sayre (SKS) e oftalmoplegia externa progressiva crônica (OEPC)
A SKS acomete pacientes com menos de 20 anos de idade e causa oftalmoplegia, ptose, retinite pigmentosa e enfraquecimento miopático. É comum a ocorrência de baixa estatura, defeitos de condução cardíaca, níveis elevados de proteína no líquido cerebrospinal, síndromes cerebelares, perda auditiva sensorioneural e níveis séricos de lactato elevados. O exame de biópsia de músculo revela a presença de fibras vermelhas esfarrapadas. Em indivíduos com mais de 20 anos de idade com fenótipo predominante de oftalmoplegia, a SKS é classificada como oftalmoplegia externa progressiva crônica (OEPC).62-65
A SKS e a OEPC mais restrita são caracterizadas por uma deleção ampla e única no mtDNA, envolvendo 5 a 50 pares de bases. Mais frequentemente, a deleção ocorre de forma esporádica e raramente é herdada da mãe. A FOX é defeituosa, e os níveis de atividade dos complexos I e IV estão reduzidos. As variantes de OEPC, que são caracterizadas por múltiplas deleções junto ao mtDNA envolvendo genes de FOX codificados no DNA nuclear, podem ser transmitidas de modo autossômico dominante ou recessivo.
Encefalomiopatia neurogastrintestinal mitocondrial (ENGM)
Uma forma especial de OEPC autossômica recessiva consiste em uma síndrome multissistêmica conhecida como encefalomiopatia neurogastrintestinal mitocondrial (ENGM).62-65 A doença pode ocorrer em indivíduos com 20 a 60 anos de idade. Os pacientes apresentam uma oftalmoplegia externa progressiva acompanhada de disfunção intestinal, neuropatia periférica e leucoencefalopatia. Os pacientes com ENGM apresentam níveis séricos de timidina aumentados e menor atividade de timidina fosforilase nos leucócitos. Estes achados, aliados às mutações no gene codificador da timidina fosforilase nuclear, com consequente comprometimento da replicação e reparo do mtDNA, confirmam o diagnóstico.
Mutações pontuais no DNA mitocondrial (mtDNA) herdadas da mãe
Síndrome MELAS (encefalopatia mitocondrial, acidose láctica e episódios do tipo acidente vascular cerebral)
A encefalopatia mitocondrial, a acidose láctica e os episódios do tipo acidente vascular cerebral combinam-se para formar a síndrome MELAS. As biópsias de músculo obtidas de pacientes afetados revelam a presença de fibras vermelhas esfarrapadas. Estes pacientes também podem apresentar perda da audição, estatura baixa, miocardiopatia, diabetes ou degenerações pigmentares de retina, de modo semelhante ao observado na SKS ou na OEPC. Até 80% dos pacientes apresentam mutações pontuais no mtDNA envolvendo o gene da leucina, junto ao RNA transportador (tRNA). Assim como outras mutações que ocorrem no mtDNA, estas mutações são heteroplásmicas, e isto implica na coexistência das formas normal e mutante do mtDNA em uma determinada célula. Uma célula normalmente contém 2 a 10 moléculas de mtDNA, que permitem a manutenção da mutação letal (isto é, comprometimento letal da FOX) em organismos viáveis.62-65
Síndrome MERRF (epilepsia mioclônica e miopatia com fibras vermelhas esfarrapadas)
A síndrome MERRF consiste na epilepsia mioclônica e miopatia com fibras vermelhas esfarrapadas. Além disso, também é comum haver ataxia, demência, surdez, enfraquecimento, desgaste e anormalidades cardíacas, embora a expressão destas condições seja variável e dependa do grau de heteroplasmia. Cerca de 80% dos pacientes com MERRF apresentam uma mutação no gene da lisina ao nível do tRNA.62-65
Neuropatia óptica hereditária de Leber (NOHL)
A NOHL representa a causa mais comum de cegueira entre adultos jovens (a prevalência é maior entre os homens do que entre as mulheres). Os pacientes apresentam perda visual indolor, subaguda e bilateral. Em pelo menos 90% das famílias, são observadas várias mutações no mtDNA. Alguns pacientes com NOHL e mutações distintas podem ter outras condições associadas, como encefalopatia, surdez, ataxia, mielopatia ou distonia.62-65
Síndrome da depleção do DNA mitocondrial (mtDNA)
Trata-se de um distúrbio autossômico recessivo, que geralmente é fatal ao redor dos 3 anos de idade, embora tenha sido encontrado em pacientes mais velhos. Afeta a musculatura, fígado, rins e cérebro. A morte é causada pela encefalopatia ou insuficiência respiratória. O defeito mitocondrial é quantitativo, em vez de qualitativo. O distúrbio resulta de desequilíbrios junto ao pool nucleotídico que, por sua vez, comprometem a replicação e o reparo do mtDNA.62-65
O tratamento de todos os distúrbios mitocondriais é suportivo e sintomático. Frequentemente, utiliza-se coenzima Q10, creatinina, carnitina e vitaminas. A prática de exercícios moderados é recomendada.
Patologias de canais iônicos, paralisias periódicas e miotonias não distróficas
As patologias de canais iônicos que afetam a excitabilidade das fibras musculares produzem uma gama de distúrbios, dentre os quais a paralisia periódica, a miotonia e a ataxia-mioquimia episódica.66-68Estas condições representam um grupo raro de distúrbios que geralmente surgem durante a infância e, de forma típica, se manifestam como ataques de paralisia. Durante os ataques paralíticos, é comum haver alterações dos níveis séricos de potássio. A miotonia também é comum em algumas formas. A suspeita destes distúrbios é levantada diante de uma história de ataques similares em familiares do paciente, sobretudo quando esses ataques são provocados pelo repouso subsequente à prática de exercícios ou por certas refeições enriquecidas com carboidratos. O diagnóstico é confirmado por análise genética do DNA sanguíneo.
Após a excitação ao nível da junção neuromuscular, os potenciais de ação são propagados por fluxos iônicos ao longo da membrana do sarcolema, que dependem da abertura (ativação) e do fechamento (inativação) do canal iônico apropriado. Nas células musculares e nervosas, a abertura dos canais de Na+ controlados por voltagem resulta em um rápido aumento da permeabilidade ao Na+ e consequente despolarização da membrana. Entretanto, para a membrana iniciar o próximo potencial de ação, é necessário que ocorra o fechamento dos canais de Na+.66-69 Os canais de potássio controlados por voltagem (CKCV) abrem-se, e os íons K+ fluem para fora da célula, criando uma voltagem hiperpolarizada ao longo da membrana celular. Os canais de Cl– contribuem para a repolarização, estabilizando o potencial de membrana.
As perturbações que afetam a estabilidade da membrana podem acarretar miotonia com ou sem paralisias periódicas. A miotonia, como sintoma manifestado nas miotonias não distróficas, ocorre nos distúrbios de canais de sódio ou de cloreto. A miotonia manifesta-se como uma rigidez indolor subsequente a um período de inatividade, que melhora após a realização de movimentos contínuos (fenômeno do aquecimento). A miotonia que se desenvolve após a exposição ao frio e piora com o exercício é denominada miotonia paradoxal. A outra manifestação de excitabilidade da membrana é a paralisia periódica, que se caracteriza por ataques paralíticos associados à hipercalemia (decorrente de mutações nos genes codificadores do canal de Na+) ou hipocalemia (causada por mutações no gene codificador do canal de cálcio voltagem-dependente). Os pacientes com paralisia periódica cardiodisrítmica (síndrome de Andersen) e aqueles com ataxia-mioquimia episódica apresentam mutações no gene codificador do canal de potássio.67-69
Distúrbios do canal de sódio
Na musculatura esquelética, os distúrbios do canal de sódio resultam de mutações em SCN4A, o gene codificador do canal de sódio. As mutações neste gene podem produzir os fenótipos clínicos de paralisia periódica hiper e normocalêmica (PP-hiper-C), paramiotonia congênita e miotonia agravada por potássio (antiga miotonia acetazolamida-responsiva ou miotonia flutuante). Os pacientes com estes distúrbios alélicos apresentam graus variáveis de miotonia de fechamento do olho, mastigação, deglutição e movimentos preensores das mãos.
A PP-hiper-C e a paramiotonia congênita (também chamada miotonia paradoxal) são doenças autossômicas dominantes. Ambas são caracterizadas por ataques de enfraquecimento que surgem durante o 1º ano de vida ou no início da infância. Os ataques de PP-hiper-C são precipitados por repouso após o exercício, administração de K+, estresse, frio e certos alimentos. Os pacientes podem apresentar miotonia leve, e é comum haver miotonia paradoxal palpebral. Os pacientes com paramiotonia congênita desenvolvem uma miotonia paradoxal que piora com a prática repetida de exercícios. Quando expostos ao frio, estes pacientes desenvolvem rigidez, especialmente na face, língua, pálpebras e mãos. Os ataques episódicos de enfraquecimento também são comuns, assemelham-se àqueles observados na PP-hiper-C e são acompanhados de miotonia. Os sintomas apresentados pelos pacientes com paramiotonia congênita, incluindo a frequência dos ataques e o enfraquecimento interataques, são tratados com inibidores de anidrase carbônica (p. ex., acetazolamida e diclorfenamida).67-69
Distúrbios do canal de potássio
As mutações em ponto no gene codificador de canal de potássio KCNA1, localizado no cromossomo 12, foram associadas à ataxia-mioquimia episódica. As mutações em outros genes codificadores de canal de potássio resultam na síndrome do QT longo. A síndrome de Andersen é causada por mutações no gene de canal de potássio KCNJ2, que codifica o gene do canal de K+ interno-retificante, Kir21, localizado no cromossomo 17q. A síndrome de Andersen é caracterizada por: paralisia periódica que pode ser acompanhada de enfraquecimento fixo; síndrome do QT longo com arritmias cardíacas ventriculares; e aspectos craniofaciais dismórficos (p. ex., micrognatia, orelhas baixas, estatura baixa e sindactilia). A síndrome de Andersen, bem como outros distúrbios do canal de potássio, geralmente responde aos inibidores de anidrase carbônica.67-69
Distúrbios do canal de cálcio
Os distúrbios do canal de cálcio manifestam-se como paralisia periódica hipocalêmica (PP-hipo-C). Trata-se de uma doença autossômica dominante, cujo gene determinante está localizado no cromossomo 1q31-q32. É causada por mutações no gene do canal de cálcio do tipo L di-hidropiridina-sensível.67-70 A PP-hipo-C manifesta-se como ataques paralíticos episódicos que surgem na 1ª ou 2ª década da vida, geralmente durante o sono. Os ataques são precipitados pelo consumo de grandes quantidades de carboidrato, repouso após o exercício e estados de excitação. Durante os ataques, a concentração sérica de K+ diminui e ocorre retenção urinária de Na+ e água. Entre os ataques e em fases tardias da vida, muitos pacientes desenvolvem uma miopatia de evolução lenta e permanente.
A doença deve ser distinguida de todas as causas de hipocalemia secundária que podem acarretar enfraquecimento, incluindo o uso de diuréticos, doença renal, hiperaldosteronismo, intoxicação por alcaçuz, uso abusivo de laxantes e doenças gastrintestinais perdedoras de potássio. Nas hipocalemias secundárias, os ataques paralíticos somente ocorrem quando a concentração de K+ cai para menos de 3 mEq (em geral, abaixo de 2,5 mEq). Em contraste, na PP-hipo-C, os ataques ocorrem até mesmo diante de concentrações séricas de K+ quase normais.
A PP-hipo-C pode estar associada à tireotoxicose, especialmente em descendentes de asiáticos. Nestes casos, a PP-hipo-C responde ao propranolol ou é resolvida após a reversão do paciente para o estado eutireoide. Embora a PP-hipo-C seja herdada de modo dominante, 1/3 dos pacientes podem apresentar doença esporádica. A frequência dos ataques e o enfraquecimento interataques são responsivos ao inibidor de anidrase carbônica acetazolamina e à diclorfenamida.71,72
Distúrbios do canal de cloreto
Os distúrbios do canal de cloreto incluem a miotonia congênita dominante (doença de Tomsen), que ocorre na 1ª década da vida e com frequência está associada à hipertrofia muscular, bem como a miotonia congênita autossômica recessiva (miotonia do tipo Becker [DMB]), que surge tardiamente na vida e pode ser mais severa.67,68 Ambas as doenças foram ligadas à ocorrência de mutações no gene do canal de cloreto, que está localizado no cromossomo 7q32 e é denominado locus CLCN1.66-68 Os pacientes que sofrem deste distúrbio não apresentam paralisia periódica nem enfraquecimento, embora às vezes possa haver um enfraquecimento severo na miotonia do tipo Becker. Este tipo de enfraquecimento melhora com a prática de exercícios. A típica miotonia de percussão e a miotonia generalizada, que se manifesta nos pacientes como rigidez e hipertrofia das pernas e nádegas, são achados característicos. A rigidez miotônica melhora com a prática de exercícios (fenômeno do aquecimento). Os sintomas miotônicos podem responder de modo variável à fenitoína, mexiletina ou tocainida.
Neuromiotonia isolada (doença de Isaac)
A neuromiotonia adquirida autoimune ocorre como neuromiotonia isolada (também chamada de doença de Isaac) e sob a forma de neuromiotonia associada à neuropatia, miastenia grave (MG) e timoma. A forma isolada da doença é caracterizada por: (1) mioquimia, que consiste no espasmo de músculos ondulados durante o repouso; (2) comprometimento do relaxamento e rigidez muscular em repouso; (3) cãibras dolorosas; e (4) sudorese aumentada. É causada pela hiperatividade das terminações nervosas motoras periféricas e atualmente é denominada neuromiotonia porque a atividade contínua da fibra muscular é abolida pelo curare, e não pelo bloqueio do nervo proximal.73,74 A neuromiotonia ocorre esporadicamente, porém também há relatos de casos familiares.73,74A EMG confirma a presença de neuromiotonia, e a determinação da concentração de autoanticorpos contra o canal de K+ identifica a natureza autoimune da doença.
O tratamento é sintomático (são usadas fenitoína, carbamazepina e mexiletina). Os casos resistentes podem necessitar de imunoterapia com imunoglobulina endovenosa (IVIg) ou plasmaférese.
Miopatias fármaco-induzidas
Embora as miopatias fármaco-induzidas sejam comuns, seu diagnóstico pode ser enganoso na prática clínica. A miopatia induzida por fármacos é definida como sendo uma manifestação subaguda e raramente aguda dos sintomas miopáticos, entre os quais o enfraquecimento muscular, fadiga, mialgia, elevação de CK ou mioglobinúria, que ocorrem em pacientes sem doença muscular diante da exposição a doses terapêuticas de certos fármacos.75-77 Após a descontinuação do agente suspeito, os sinais clínicos ou bioquímicos de envolvimento muscular geralmente melhoram, sustentando o efeito causal do fármaco miotóxico agressor. Entretanto, há casos em que a miocitotoxicidade é irreversível, como se observa com alguns análogos de nucleosídeo que são incorporados à cadeia do mtDNA.78-80 O exame de biópsia de músculo é essencial para fins de documentação da miotoxicidade, mas às vezes pode não ser informativo, como ocorre no caso de alguns pacientes com mioglobinúria ou enfraquecimento muscular leve decorrente do uso de estatinas ou análogos de nucleosídeo.75-77
Os agentes miotóxicos podem causar miopatia por meio de alguns mecanismos: ao afetarem diretamente uma organela muscular (p. ex., mitocôndria, os lisossomos, microtúbulos ou filamentos espessos);75-77 alterando antígenos musculares e, assim, induzindo uma reação inflamatória imunológica; ou ao induzirem certos efeitos sistêmicos secundários, como perturbação eletrolítica, deprivação nutricional (p. ex., quando o agente compete com as vitaminas) e má absorção. Vários fármacos podem ter ação miotóxica quando usados de modo isolado ou combinados a outros fármacos. Além de saber quais fármacos são miotóxicos, os clínicos devem sempre estar alertas quanto ao potencial de miotoxicidade dos novos fármacos lançados no mercado. Os fármacos que mais comumente induzem toxicidade muscular são descritos a seguir.
Agentes redutores de colesterol: miopatias induzidas por estatinas
Vários fármacos redutores de colesterol causam uma miopatia reversível que é caracterizada por enfraquecimento da musculatura proximal, mialgia, elevação dos níveis séricos de CK e alterações miopáticas de EMG, incluindo potenciais de fibrilação e descargas repetitivas complexas ou miotônicas. A biópsia de músculo pode apresentar alterações mínimas e inespecíficas, bem como a presença ocasional de fibras necróticas ou vermelhas esfarrapadas.76,77,81,82 A recuperação gradual ocorre após a suspensão do fármaco agressor.
Como o colesterol é o principal esterol constituinte das membranas musculares, a diminuição do pool de colesterol normal disponível para síntese de membrana pode aumentar a fluidez desta estrutura, acarretando a instabilidade do sarcolema, descargas miotônicas e, em casos avançados, rabdomiólise.76,77,81,82 As estatinas também inibem a produção de ubiquinona e, desta forma, interferem no metabolismo de energia e ATP mitocondrial do miócito. Além disso, as estatinas podem induzir alterações nas células T imunorregulatórias e nas moléculas de adesão, bem como desencadear uma miopatia imunomediada com regulação positiva do MHC-I.76
As estatinas podem produzir os seguintes sinais e sintomas miopáticos:76 (a) elevação da CK (até 10 vezes o valor normal) assintomática e flutuante em até 5% dos pacientes; (b) mialgia, com ou sem elevação da K, em até 9 a 25% dos pacientes; (c) enfraquecimento muscular leve a moderado com elevação da CK atribuível a uma miopatia tóxica ou a uma miopatia necrotizante autoimune responsiva a imunoterapias (em alguns casos, o uso de estatinas revela uma condição miopática preexistente); e (d) rabdomiólise, definida pela elevação aguda dos níveis de CK (> 15.000 em relação ao limite superior normal) frequentemente acompanhada de mialgia, enfraquecimento e mioglobinúria. A incidência de mioglobinúria ocorre em menos de 1 a cada 100.000 prescrições e varia de 0 para fluvastatina; 0,04 para pravastatina e atorvastatina; 0,19 para lovastatina; até 3,16 para cervistatina (que foi retirada do mercado).76,77,81,82 A coadministração de ciclosporina e lovastatina a pacientes com transplante de coração ou rim e hiperlipidemia aumenta a incidência de miopatia com rabdomiólise. A cerivastatina era a estatina mais comumente associada aos sintomas miopáticos e, subsequentemente, foi sucedida pela lovastatina, fluvastatina, atorvastatina, sinvastatina e pravastatina.81,82 O risco de miopatia induzida por estatina aumenta com o uso de doses mais altas, estatinas com ação lipofílica (incluindo todas, exceto a pravastatina e a rosuvastatina) e com a terapia concomitante com fármacos como gemfibrozil, amiodarona, colchicina ou ciclosporina.81,82
Os sintomas miopáticos geralmente melhoram com a interrupção do uso destes fármacos. Quando os sintomas persistem ou continuam a piorar, torna-se necessário instituir uma investigação maior com exame de biópsia de músculo para excluir a hipótese de miopatia necrotizante imune (uma condição que requer imunoterapia com esteroides ou imunoglobulina IV) ou outra condição miopática previamente não identificada. O valor da coenzima Q10 no tratamento dos sintomas miopáticos ainda é incerto.81,82
Miopatia induzida por fármacos antirreumáticos, anti-inflamatórios e imunossupressores
Esta categoria inclui os seguintes fármacos:
1. D-penicilamina – o mais bem conhecido dos fármacos responsáveis pelas complicações neuromusculares imunomediadas, entre as quais a polimiosite e a miastenia, observadas em cerca de 0,6% dos pacientes tratados.
2. Cloroquina e hidroxicloroquina – efeitos observados com a administração prolongada de altas doses de cloroquina (500 mg/dia). Clínica e histologicamente, esta miopatia assemelha-se à DMA e é caracterizada por um enfraquecimento muscular que muitas vezes é acompanhado de níveis normais de CK e presença de múltiplos vacúolos contendo material positivo para fosfatase ácida atribuíveis à atividade lisossômica aumentada.83 A miopatia é lentamente revertida pela descontinuação do fármaco.
3. Colchicina – interfere no crescimento dos microtúbulos. Seu uso prolongado acarreta uma miopatia vacuolar com acúmulo de lisossomos e vacúolos autofágicos. Os sintomas incluem enfraquecimento da musculatura proximal, elevação dos níveis séricos de CK, envolvimento sensorial distal e arreflexia. Estes sintomas são resolvidos em 4 a 6 semanas após a suspensão do uso do fármaco.
4. Ciclosporina e tacrolimo – em nossa própria experiência, é bastante raro estes fármacos causarem miopatia por si só, embora estejam implicados na miotoxicidade quando são usados de modo concomitante com as estatinas ou a colchicina.75-77
Miopatia com o uso de zidovudina (AZT) e outros análogos de nucleosídeos
A miopatia induzida pelo AZT manifesta-se como enfraquecimento da musculatura proximal, mialgia (de modo predominante nas coxas e panturrilhas), fadiga, alterações miopáticas na EMG e níveis séricos de CK elevados.61,77-79 Os sintomas são resolvidos em 4 a 6 semanas após a suspensão do uso de AZT.78,79 O aspecto histológico exclusivo da miopatia induzida por AZT consiste na presença de fibras vermelhas esfarrapadas contendo acúmulos lipídicos e de numerosas fibras negativas para citocromo-c oxidase, sugestivas de anormalidades mitocondriais.61,78,79 O AZT é um finalizador da cadeia de DNA que inibe a gama-DNA polimerase na matriz mitocondrial. Desta forma, há síntese de mtDNA e, como resultado, até 78% do mtDNA muscular é depletado.78,79 Entre outros análogos de nucleosídeos utilizados no tratamento da Aids, a estavudina também pode ser miotóxica. A síndrome de lipodistrofia, acidose láctica e miopatia tem sido observada na terapia antirretroviral altamente ativa com o uso de um dos inibidores de protease recém-lançados combinado a dois análogos de nucleosídeos, em especial a estavudina (d4T).80
Fármacos contaminantes que causam predominantemente fasciite
O uso de L-triptofano contaminado foi responsável por uma epidemia de uma síndrome de fasciíte eosinofílica, miosite, espessamento cutâneo, neuropatia axonal e outras manifestações sistêmicas. Um processo imunológico dirigido contra os fibroblastos da matriz extracelular, deflagrado pelo contaminante, foi implicado como causa desta síndrome (denominada síndrome da eosinofilia-mialgia).84 Uma síndrome similar atribuível ao óleo contaminado (síndrome do óleo tóxico) ocorreu na Espanha, há muitos anos. A miofasciite macrofágica é uma entidade encontrada na França, que resulta do uso de vacinas contendo alumínio. O exame da biópsia obtida dos sítios de inoculação das vacinas revela o acúmulo de macrófagos no epimísio, perimísio e endomísio.
Miopatia associada a doenças graves
Em pacientes com paralisia prolongada induzida por agentes bloqueadores não despolarizantes (p. ex., pancurônio), pode haver desenvolvimento de miopatia aguda após a suspensão da ventilação mecânica. A maioria destes pacientes recebeu doses altas de corticosteroides para tratamento de condição asmática ou de outra doença sistêmica, em que uma paralisia artificial tenha sido induzida para assegurar uma toalete pulmonar agressiva.76,77,85,86 O uso combinado de agentes bloqueadores e corticosteroides foi implicado de forma consistente no desenvolvimento de miopatia severa, porém os pacientes às vezes são apenas minimamente expostos a 1 destes 2 agentes.77,85-87
A manifestação clínica é caracterizada pelo enfraquecimento generalizado e severo de todos os membros, falha no desmame da ventilação mecânica, atrofia muscular, níveis séricos de CK normais ou moderadamente elevados e alterações miopáticas na EMG. O enfraquecimento costuma melhorar lentamente.77,85-87 A miopatia induzida por agente bloqueador-corticosteroide pode coexistir com uma neuropatia axonal. Entretanto, segundo o consenso atual, a doença paralítica é causada predominantemente por uma miopatia. O diagnóstico diferencial deve incluir defeitos de transmissão neuromuscular, como MG, síndrome de Guillain-Barré, miopatia necrotizante aguda ou inflamatória e paralisia periódica. A biópsia muscular mostra anormalidades morfológicas extensas, que resultam da perda seletiva de filamentos espessos sem sinais de necrose, inflamação ou fagocitose. Com a coloração de adenosina trifosfatase (ATPse), são observadas áreas claras centrais notáveis em muitas fibras.77,86,87 A expressão de calpaína aumenta acentuadamente, implicando na alteração da homeostasia do cálcio e intensificação da proteólise. A microscopia eletrônica revela uma perda seletiva e extensiva de miofilamentos espessos. Os miofilamentos delgados e os discos Z são preservados.
Altas doses de esteroides devem ser usadas com cautela por pacientes sob tratamento prolongado com agentes paralíticos. A miopatia melhora lentamente com a reabilitação intensa.
Miopatia induzida por esteroide
Os pacientes com hiperadrenocorticismo (isto é, síndrome de Cushing) podem desenvolver enfraquecimento. Alterações similares podem ocorrer durante a administração prolongada de prednisona (em geral, dosagens acima de 20 mg/dia) ou dexametasona, especialmente em pacientes que apresentam movimentação precária. O enfraquecimento miopático induzido por esteroides é leve, poupa os músculos flexores do pescoço77 e pode agravar o enfraquecimento causado pela doença imune ou malignidade subjacente para a qual os esteroides estejam sendo administrados. A diminuição da dose do fármaco reverte a miopatia. Os níveis séricos de CK permanecem normais, o exame da biópsia de músculo mostra apenas uma atrofia de fibras do tipo II, e a EMG não é informativo.76,77
Toxicidade por drogas ilícitas
Entre as drogas ilícitas estão a cocaína, heroína, anfetaminas, feniciclidina (PCP) e o etanol. Estas substâncias causam rabdomiólise e, eventualmente, síndrome da compressão. As drogas ilícitas quase sempre são misturadas a vários agentes. Ainda é preciso determinar se as drogas afetam a musculatura por si só ou se é a combinação das drogas com os agentes adulterantes que deflagra a toxicidade.76,77
Os pacientes alcoólatras podem desenvolver uma miopatia aguda ou crônica. A miopatia aguda manifesta-se como rabdomiólise ou mioglobinúria e é precedida por edema e dor muscular. A condição pode recorrer se o paciente voltar a beber. A miopatia aguda em pacientes com alcoolismo também pode estar relacionada à hipocalemia, quando a concentração sérica de K+ cai para menos de 2,5 mEq. Esta miopatia é indolor, não é acompanhada de inchaço muscular e é rapidamente reversível. O enfraquecimento da musculatura proximal em pacientes com alcoolismo por tempo prolongado muitas vezes é multifatorial e não necessariamente atribuível a um processo miopático primário. Outros fatores que também podem estar envolvidos são a desnutrição, inatividade ou doença neurogênica. Em termos de histologia, a atrofia das fibras de tipo II constitui a anormalidade mais comum. Alguns indivíduos consumidores de bebidas alcoólicas por tempo prolongado podem apresentar uma elevação assintomática dos níveis séricos de CK – até 20 vezes acima dos níveis normais – que é agravada pela atividade física.
Embora as causas exógenas de mioglobinúria possam ser multifatoriais, as drogas ilícitas e o álcool podem ser responsáveis pela maioria dos casos. Em uma das maiores séries recentes, envolvendo 475 pacientes, a causa era o uso de drogas ilícitas ou o alcoolismo em 163 pacientes, seguidas do uso de fármacos (estatinas, colchicina, AZT, lítio) em 54 pacientes.88 As causas menos frequentes foram as doenças musculares (49), traumatismo (42), síndromes malignas neurolépticas (38), causas idiopáticas (34), convulsões (32), imobilidade (21) e condições médicas diversas (nos 40 pacientes restantes).
Distúrbios da transmissão neuromuscular
As principais áreas da junção neuromuscular são: (1) a região pré-sináptica, com zonas ativas contendo linhas duplas paralelas de canais de cálcio controladas por voltagem (CCCV) e vesículas sinápticas contendo acetilcolina (ACh); (2) o espaço sináptico; e (3) a região pós-sináptica, constituída por dobras juncionais contendo receptores de acetilcolina (AChRs) [Figura 3].73,74,89,90 Ao nível funcional, a transmissão neuromuscular depende da liberação de ACh pelo nervo motor terminal, da interação da ACh com AChR e da resultante despolarização da fibra muscular. No estado de repouso, há liberação aleatória de ACh a partir de uma única vesícula. A ACh liga-se ao AChR e abre seu canal iônico cátion-seletivo, permitindo a entrada de Na+ na região da placa terminal. Isto cria um potencial de despolarização denominado potencial de placa terminal (PPT) em miniatura.73,74,89,90 A ACh então é dissociada do AChR e rapidamente hidrolisada pela acetilcolinesterase (AChE). Quando a AChE é inibida, como se observa no tratamento com fármacos contendo AChE (p. ex., piridostigmina), mais moléculas ACh se ligam ao AChR e abrem múltiplos canais iônicos.
Figura 3. A transmissão a partir da terminação nervosa na junção neuromuscular ocorre em sítios localizados (zonas ativas). A região pré-sináptica possui canais de cálcio controlados por voltagem (CCCV) e vesículas sinápticas que contêm 6.000 a 8.000 moléculas do neurotransmissor acetilcolina (ACh). Quando o potencial de ação chega na terminação nervosa, os CCCV abrem-se, e o Ca2+ flui para dentro do terminal nervoso. A entrada de Ca2+ deflagra a fusão e exocitose das vesículas sinápticas. A ACh é liberada dentro do espaço sináptico e liga-se ao receptor de acetilcolina (AChR) localizado na região da placa terminal da membrana muscular (dobras pós-sinápticas). A ativação do AChR gera um fluxo de íons pelo AChR, atravessando a membrana, que inicia a resposta elétrica muscular denominada potencial de placa terminal (PPT). O PPT dissemina-se da região da placa terminal para a membrana muscular circundante e inicia a resposta de impulso do músculo que, por fim, produz a contração muscular. Os canais de potássio controlados por voltagem (CKCV) são canais transmembrana específicos para potássio e sensíveis às alterações de voltagem associadas ao potencial de membrana celular. Estes canais exercem papel essencial no processo de trazer a célula despolarizada de volta ao estado de repouso. Os anticorpos dirigidos contra os CKCV são responsáveis pela neuromiotonia.
Quando o potencial de ação chega ao terminal nervoso, o CCCV abre e os íons de Ca2+ fluem para dentro do terminal nervoso. A entrada de Ca2+ deflagra a fusão e exocitose de vesículas sinápticas, bem como a liberação de moléculas de ACh (cada vesícula contém 6.000 a 10.000 moléculas de ACh) que se difundem e disseminam sobre as dobras juncionais. A liberação simultânea de ACh pela fusão de 50 a 300 vesículas resulta em PPT. Se o PPT ultrapassar determinado limiar, um potencial de ação muscular (PAM) desencadeia a contração muscular. Normalmente, as interações entre a ACh liberada e o AChR são mais do que suficientes para produzir um PPT capaz de deflagrar um PAM. A diferença entre a amplitude real do PPT e a amplitude de PPT necessária para deflagrar o PAM é denominada margem de segurança.
Existem 3 distúrbios principais que afetam a transmissão neuromuscular: MG, síndrome miastênica de Lambert-Eaton (SMLE) e várias síndromes miastênicas congênitas.
Miastenia grave (MG)
A MG é um protótipo de doença autoimune que atende a todos os critérios imunológicos:73,74,89,90 o antígeno é o AChR; há anticorpos dirigidos contra o AChR que são detectados e quantificados no soro dos pacientes afetados; a doença pode ser transmitida aos animais pela IgG patogênica do paciente; a imunoglobulina sérica liga-se ao AChR e causa a destruição dos receptores via fixação do complemento, afetando a transmissão neuromuscular; e a remoção dos anticorpos resulta em melhora clínica.
Na MG, os anticorpos dirigidos contra o AChR diminuem o número de receptores disponíveis. Em consequência, a ACh liberada não tem como se ligar ao número necessário de receptores. Desta forma, o PPT geral é insuficiente para deflagrar um PAM e a transmissão neuromuscular falha. Quando esta falha ocorre em muitas junções neuromusculares, o músculo não pode ser totalmente ativado e o resultado é a fadiga e o enfraquecimento muscular. Durante a estimulação repetitiva do nervo em indivíduos saudáveis, a quantidade de ACh liberada é reduzida após os primeiros impulsos, porque o nervo não consegue sustentar sua frequência original de liberação. Entretanto, a transmissão neuromuscular não falha, pois a margem de segurança é suficiente. Em pacientes com MG, o PPT já é pequeno. Durante a estimulação repetida do nervo, o PTT é deprimido ainda mais porque as quantidades decrescentes de ACh liberadas por impulso subsequente são insuficientes para ligar o número reduzido de AChRs. Isto resulta em um decréscimo adicional do PAM. Os exames de estimulação repetida de nervo são utilizados com fins diagnósticos, para demonstrar o comprometimento da transmissão neuromuscular em pacientes com MG. Como a MG afeta o AChR nicotínico na junção neuromuscular, apenas os sistemas motores são afetados.
Epidemiologia
A prevalência da MG é de aproximadamente 4 a 6 casos para cada 100.000 pacientes. A incidência anual varia, mas são identificados dois picos: um pico está associado às mulheres na 2ª ou 3ª década da vida, enquanto o outro pico está associado sobretudo aos homens com mais de 60 anos de idade. Há um relato recente de MG subdiagnosticada em pacientes com mais de 70 anos de idade.91 A MG clássica autoimune também pode se desenvolver durante a infância e responde bem à timectomia ou ao uso de anticolinesterases. A MG pode estar associada ao timoma em até 15% dos pacientes adultos ou pode se desenvolver após a remoção de um timoma. Além disso, até 5% dos pacientes com MG apresentam outras condições autoimunes sistêmicas, até 15% dos pacientes apresentam diversos autoanticorpos, até 20% dos pacientes possuem doença da tireoide, e até 5% dos pacientes têm história familiar de outra doença autoimune. A gestação produz um efeito variável, mas na maioria das pacientes o enfraquecimento aumenta durante o período pós-parto. Cerca de 12% das crianças nascidas de mães com MG desenvolvem MG neonatal transiente, como resultado da transferência transplacentária de anticorpos anti-AChR circulantes da mão miastênica para o feto. Estes bebês desenvolvem enfraquecimento, dificuldades de alimentação, problemas respiratórios, um choro fraco e enfraquecimento facial ao longo das primeiras horas após o nascimento. A condição dura até 3 semanas, coincidindo com o tempo de meia-vida da IgG.73,74,89,90
Imunopatogênese
A MG é mediada por autoanticorpos patogênicos que se ligam ao AChR, acarretando uma perda funcional de receptores e a destruição focal das dobras juncionais pós-sinápticas, interferindo na despolarização da membrana pós-sináptica [Figura 4].73,74,89,90Entre 10 e 15% dos pacientes com MG, especialmente aqueles com as formas branda, infantil ou localizada (p. ex., ocular), não possuem anticorpos detectáveis contra o AChR. A maioria destes pacientes, ainda que soronegativa, apresentam um distúrbio mediado por anticorpos. Entre os 15% de pacientes soronegativos, cerca de 6% apresentam anticorpos contra MuSK,92,93 uma quinase músculo-específica necessária ao desenvolvimento e agrupamento de AChR na junção neuromuscular [Figura 4]. Os restantes 9% de pacientes soronegativos possuem anticorpos IgG precariamente caracterizados, que mediam a inibição reversível da função do AChR, ou anticorpos IgM, que mediam a inibição indireta da função do AChR.94
Figura 4. Destruição do receptor de acetilcolina (AChR) na miastenia grave (MG), por ação de autoanticorpos. Os autoanticorpos anti-AChR são patogênicos e comprometem a transmissão neuromuscular (1) ao bloquearem os AchR disponíveis, prevenindo, assim, a ligação da acetilcolina (ACh) ao seu receptor (o significado funcional deste efeito é considerado mínimo); (2) ao realizarem ligação cruzada com os AChR e, deste modo, acelerarem a internalização e degradação proteolítica do AChR via mecanismo de modulação antigênica; e por meio da fixação de complemento, em um processo que leva (3) deposição do complexo de ataque à membrana (CAM) lítico junto aos AChR, resultando (4) na lise progressiva da membrana e liberação das dobras pós-sinápticas no espaço sináptico. Estes processos levam à simplificação da membrana pós-sináptica e à perda de AChR. Cerca de 6% dos pacientes sem anticorpos anti-AChR possuem anticorpos dirigidos contra MuSK, uma quinase músculo-específica necessária ao desenvolvimento e agrupamento dos AChR. O papel patogênico dos anticorpos antiMuSK ainda é desconhecido.
CCCV = canais de cálcio controlados por voltagem; CKCV = canais de potássiocontrolados por voltagem.
Outros anticorpos associados à MG são os anticorpos anticélula estriada. Estes anticorpos são encontrados em até 80% dos pacientes com timoma e MG, em 25% dos pacientes com timoma sem MG, em 30% dos pacientes adultos com MG (55% dos casos com início da MG após os 60 anos de idade), em 6% dos pacientes com SMLE, e em 3% dos pacientes com câncer de pulmão e doença hepática autoimune.94 Os anticorpos antititina também estão presentes em 55% dos pacientes AChR-positivos com MG de início tardio, independentemente da existência de um timoma. Os anticorpos dirigidos contra os microssomos tireóideos e contra a tireoglobulina são encontrados com frequência em pacientes que apresentam MG ocular, bem como em pacientes com SMLE sem câncer.
O AChR é um antígeno dependente de célula T. Na MG, as células CD4+ são sensibilizadas e respondem à estimulação com AChR ou aos peptídeos imunodominantes sintéticos de AChR.89-92 Como os linfócitos de indivíduos sadios normais também respondem a estes peptídeos, ainda que em menor grau, as células T AChR-específicas fazem parte do repertório imune normal. Isto sugere que a MG é uma doença em que a imunorregulação está anormal.
Cerca de 75% dos pacientes com MG apresentam anormalidades tímicas, seja hiperplasia com formação de centro germinativo ou timoma (15% de todos os pacientes com MG). As evidências que implicam o timo na MG baseiam-se nos seguintes fatores: (1) o timo de um indivíduo afetado contém uma população maior de células B que poderiam secretar anticorpos anti-AChR, em comparação ao timo de um indivíduo normal sadio; (2) o timo de um paciente afetado possui células mieloides contendo AChR proximais às células dendríticas interdigitantes; e (3) o número de células T sensibilizadas pelo AChR é maior no timo de um indivíduo afetado do no timo de um indivíduo saudável.
Na MG, a quebra da autotolerância pode ter início no timo. O AChR presente nas células mieloides pode, então, ser reconhecido pelas células dendríticas tímicas. Estas apresentam o antígeno às células CD4+ que, por sua vez, estimulam as células B produtoras de anticorpo a fazerem anticorpos anti-AChR.89-92 Subsequentemente, estes anticorpos reconhecem e estabelecem reações cruzadas com o AChR presente na musculatura esquelética. Acredita-se que a remoção do timo resulte em melhora clínica ou remissão, contudo o papel da timectomia atualmente está sendo investigado por um estudo controlado.
A MG frequentemente está associada a outras condições autoimunes, incluindo as doenças vasculares colágenas, polimiosite, pênfigo, doenças tireóideas autoimunes e doença do enxerto vs. hospedeiro. O uso de D-penicilamina induz a MG clássica, que melhora com a descontinuação do fármaco.
Diagnóstico
Os pacientes com MG apresentam enfraquecimento da musculatura esquelética e fadigabilidade. O enfraquecimento muitas vezes afeta a elevação das pálpebras e a movimentação dos músculos extraoculares, que resulta em diplopia e ptose assimétrica característica. Os extensores do pescoço com frequência sofrem enfraquecimento e o paciente apresenta queda da cabeça. Os pacientes com MG podem apresentar um enfraquecimento da musculatura facial e bulbar que resulta em uma aparência facial ríspida (como a de um rosnado) quando o paciente sorri; fala anasalada ou disártrica e disfônica baixa; e disfagia, que pode causar regurgitação e engasgos. A fadiga e o enfraquecimento muscular esquelético proximal geram dificuldade para caminhar, subir escadas, pentear o cabelo e carregar objetos. O enfraquecimento da muscular respiratória pode ser significativo. Os sintomas flutuam e com frequência são mais amenos de manhã. A fadigabilidade geralmente piora ao fim do dia e após realizar atividades repetidas ou contínuas. Se um paciente é solicitado a manter os braços abduzidos, observa-se o declínio gradual da altura dos braços. Se o paciente é solicitado a conversar de forma contínua, sua voz pode se tornar rouca, nasalada, indistinta e, por fim, inaudível. A sensibilidade e a cognição permanecem normais. Os reflexos tendíneos, em especial dos músculos não atróficos, tendem a ser rápidos ou normalmente ativos. Os dedos dos pés voltam-se para baixo. Os sintomas pioram durante ou antes do período menstrual e durante as infecções virais ou bacterianas.
Os pacientes com anticorpos antiMuSK tendem a ser do sexo feminino. O aparecimento da doença nestes pacientes ocorre aos 20 a 60 anos de idade. Estes pacientes frequentemente apresentam um enfraquecimento proeminente da musculatura do pescoço, ombros ou respiratória, bem como sinais bulbares.95 Outros apresentam sinais predominantemente oculobulbares.92,93
Além da demonstração do enfraquecimento fadigável durante o exame, o diagnóstico clínico da MG é confirmado pela detecção sorológica de anticorpos anti-AChR ou antiMuSK, e também pela demonstração eletrofisiológica da falha de transmissão neuromuscular. Os anticorpos anti-AChR estão presentes em até 85 a 90% dos pacientes com MG generalizada e em até 70% dos pacientes com MG ocular. Estes anticorpos também são encontrados em até 30% dos pacientes com doença hepática autoimune, 10% dos pacientes com anemia perniciosa e em até 13% dos pacientes com SMLE.95,96Em geral, os títulos de anticorpos anti-AChR não estão correlacionados com a severidade clínica. Os exames de estimulação nervosa repetitiva demonstram a ocorrência de uma rápida diminuição da amplitude (> 12%) do PAM. A EMG de fibra única mostra uma maior instabilidade de sinal e bloqueio em mais de 90% dos pacientes. Em raras circunstâncias e na maioria das vezes em casos pediátricos, realiza-se um teste com edrofônio ou neostigmina. Estes fármacos inibem a AChE e assim permitem que a ACh interaja repetidamente com os AChRs remanescentes. Como resultado, a força muscular melhora. É necessário realizar um exame de RM torácica para pesquisa de hiperplasia tímica ou timoma. A MG deve ser diferenciada das doenças ou distúrbios tóxicos induzidos por agentes exógenos que apresentam quadros clínicos similares, tais como botulismo, envenenamento por organofosfatos, reação tóxica à D-penicilamina, miopatia mitocondrial, lesão compressiva afetando nervos cranianos, SMLE e síndromes miastênicas congênitas.95,96
Tratamento
O termo “grave” atualmente é considerado uma designação incorreta, porque a MG é bastante responsiva às terapias disponíveis. Embora a sequência em que as diversas modalidades terapêuticas são aplicadas possa diferir de acordo com o clínico, a escolha inicial para os casos leves geralmente é um fármaco anticolinesterásico que é administrado a cada 4 horas, enquanto o paciente está acordado. A timectomia é uma opção para os pacientes que já passaram da puberdade e com idade até 55 anos. O efeito benéfico da timectomia, contudo, foi questionado e precisou ser avaliado por um amplo estudo multicentros que atualmente está sendo conduzido.97,98 O timoma deve ser sempre removido, com subsequente instituição de uma radioterapia.
A prednisona é o fármaco imunoterapêutico utilizado na terapia de 1ª linha. A maioria dos médicos prefere começar o tratamento com doses menores de prednisona (isto é, 20 a 40 mg/dia) e aumentar a dosagem gradualmente, para até 60 a 80 mg/dia, com o objetivo de prevenir uma piora transiente observada em alguns casos. A dose é gradualmente reduzida para um programa de dias alternados até que a menor dose capaz de controlar a doença seja alcançada. A plasmaférese ou a administração de imunoglobulina geralmente são reservadas para as crises e casos severos, bem como para fortalecer um paciente antes da timectomia.97-99 A ciclosporina, micofenolato ou azatioprina podem ser usados como terapia de 2ª linha para manter a remissão após a redução da dosagem de esteroides.97,98 O tituximabe está emergindo como terapia promissora para certos casos difíceis.100 Os pacientes com anticorpos antiMuSK não melhoram com a timectomia e sua resposta aos inibidores de colinesterase é variável. A resposta à imunoterapia geralmente é boa, ainda que variável: alguns pacientes podem desenvolver atrofia e enfraquecimento facial, faríngeo ou na língua permanente.
Síndrome miastênica de Lambert-Eaton
Na SMLE, os anticorpos específicos contra os CCCV produzem modulação antigênica e depleção dos CCCV [Figura 4]. Isto restringe o ingresso de Ca2+ no nervo motor terminal e diminui a liberação de ACh durante um impulso nervoso. A quantidade de ACh liberada pode gerar apenas um pequeno PPT, que não consegue deflagrar um PAM. Isto resulta em falha de transmissão neuromuscular. Na SMLE, uma observação importante é a associação frequente com o câncer de pequenas células do pulmão (CPCP), que também expressa CCCV.89-91,95,96 Cerca de 70% dos homens com SMLE apresentam CPCP, comparativamente aos 20% das mulheres. A SMLE pode preceder em até 3 anos a detecção de uma malignidade. A malignidade é rara em indivíduos com menos de 40 anos de idade. Acredita-se que as células tumorais deflagrem a produção de autoanticorpos dirigidos contra determinantes de superfície, que apresentam reação cruzada com epítopos similares presentes nas regiões pré-sinápticas das junções neuromusculares.
Os pacientes apresentam enfraquecimento da musculatura proximal, fadigabilidade aumentada e sintomas oculares transientes de ptose ou diplopia. A sintomatologia autonômica, em que há ressecamento da boca e dos olhos, impotência, constipação e reflexos pupilares anormais, manifesta-se com frequência. É comum haver hiporreflexia. Um sinal típico consiste no aumento da força muscular e dos reflexos decorridos alguns segundos da manutenção de um esforço de nível máximo. O diagnóstico é confirmado pela presença de anticorpos dirigidos contra os canais de cálcio, detectados em até 95% dos pacientes com SMLE 90,91,95,96 e por exames de estimulação nervosa repetida, que demonstram um aumento dramático da amplitude da PAM com a estimulação tetânica. A EMG de fibra única mostra um aumento da instabilidade de sinal e bloqueio, que melhoram com frequências de estimulação mais rápidas. Uma pesquisa para CPCP, especialmente em homens com mais de 40 anos de idade, constitui uma parte essencial da investigação do paciente.
Na SMLE associada ao CPCP, o foco primário é o tratamento do tumor, pois os sintomas melhoram conforme o câncer regride. O 1º fármaco de escolha na SMLE é a 3,4-diaminopiridina em dosagens de 40 a 60 mg/dia. Este fármaco prolonga a duração do potencial de ação pré-sináptico bloqueando as correntes de saída de K+. Isto permite a intensificação da entrada de Ca2+ no terminal nervoso e a liberação adicional de ACh. A prednisona é substancialmente útil. Outras abordagens terapêuticas adicionais são a azatioprina, plasmaférese e imunoglobulina.
Neuromiotonia com autoanticorpos anticanal de K+ (síndrome de Isaac)
A síndrome de Isaac é uma doença autoimune rara, que se manifesta como neuromiotonia isolada [ver Neuromiotonia isolada (doença de Isaac), anteriormente] ou associada a um timoma, MG e sintomas envolvendo o SNC, como encefalite límbica, transtornos da personalidade, alucinações e distúrbios do sono.73,74 Os pacientes com neuromiotonia apresentam mioquimia, miotonia e, frequentemente, hipertrofia muscular. As descargas de mioquimia e as descargas prolongadas da unidade motora são observadas ao nível eletrofisiológico. Outros pacientes desenvolvem síndrome de cãibra-fasciculação e neuropatia periférica.
Os pacientes afetados possuem anticorpos contra os CKCV, que são responsáveis pela doença.73,74 O distúrbio responde a corticosteroide, plasmaférese, IVIg ou agentes imunossupressores.
Síndromes miastênicas congênitas
As síndromes miastênicas congênitas constituem um grupo heterogêneo de síndromes que causam falha de transmissão neuromuscular não autoimune, resultante de defeitos genéticos envolvendo várias moléculas, enzimas ou canais presentes na junção neuromuscular.
A suspeita de síndrome miastênica congênita surge quando um bebê, uma criança ou, em alguns casos, um adulto jovem apresenta ptose flutuante, fadigabilidade, enfraquecimento aumentado durante o esforço contínuo (comum a todos os defeitos de transmissão neuromuscular), referenciais motores leves ou tardios, ausência de MG autoimune e história familiar positiva.101
Esclerose lateral amiotrófica (ELA)
A ELA, também conhecida como doença de Lou Gehrig, é um distúrbio neurodegenerativo de neurônios motores caracterizado pelo enfraquecimento progressivo e atrofia muscular com consequente paralisia. Classicamente, a ELA não é acompanhada de comprometimento cognitivo nem perda sensorial, embora em algumas variantes estejam presentes a demência, anomalias sensoriais ou outras alterações. A idade do paciente no momento do aparecimento da doença é variável; geralmente, ocorre na 4ª à 7ª décadas da vida. A morte por falha da musculatura respiratória tipicamente ocorre em 2 a 5 anos após o estabelecimento do diagnóstico.102 A ELA é a doença de neurônio motor do adulto mais comum, com incidência anual de 1 a 2 a cada 100.000 indivíduos, nos Estados Unidos.103 O risco de desenvolvimento da condição ao longo da vida é de aproximadamente 1 em 2.000.103
Etiologia e fatores de risco
Cerca de 5 a 10% dos casos de ELA são geneticamente determinados.104 Os demais casos (> 90%) são de natureza esporádica. As formas familiar e esporádica da doença são clinicamente indistinguíveis. Os casos familiares estão mapeados em pelos menos 12 loci e exibem diversos padrões de herança.104-107 As mutações no gene da cobre-zinco superóxido dismutase 1 (SOD1) citoplasmática, localizado no sítio 21q22.1 do cromossomo 21, são responsivas em menos de 20% dos casos hereditários.104-107 Foram descritos alelos dominantes e recessivos. O papel preciso de SOD1 na etiologia da ELA ainda é desconhecido. Existem pelo menos mais 5 loci distintos associados à ELA adulta de herança dominante, contudo os genes localizados nestes sítios não foram identificados. Outros 3 loci foram associados a variantes raras e juvenis de ELA.104,105 Em comparação com a forma esporádica da doença, a variante juvenil está associada a um curso mais lento e a uma sobrevida mais prolongada. Os pacientes com ELA juvenil tornam-se sintomáticos durante a 1ª e 2ª décadas da vida, podendo sobreviver por 20 a 30 anos após o estabelecimento do diagnóstico.105
A suscetibilidade ao desenvolvimento de ELA nos casos esporádicos foi ligada a defeitos envolvendo receptores e transportadores de glutamato na glia, acúmulo anormal de neurofilamentos nos corpos celulares e axônios proximais de neurônios motores, expressão diminuída de fator de crescimento endotelial vascular, deleções homozigotas do gene determinantes de sobrevida do neurônio motor e mutações do tipo null envolvendo o fator neurotrófico ciliar (um fator de sobrevida do neurônio motor).106,107 Acredita-se que as interações ambientais exerçam algum papel, porém nenhum fator de risco claro foi identificado. O tabagismo recentemente foi implicado como fator de risco.108
Diagnóstico
Na ELA, a perda neuronal motora seletiva ocorre junto aos cornos ventrais da medula espinal, córtex motor e na maioria dos núcleos motores do tronco encefálico. Os critérios estabelecidos pela World Federation of Neurology para o diagnóstico da ELA requerem evidências de degeneração neuronal motora superior e inferior, bem como a progressão dos sintomas junto a uma única região (isto é, tronco encefálico, medula espinal cervical, medula espinal torácica ou medula espinal lombar) ou a partir de uma região para outras.102,103 Os achados envolvendo neurônios motores superiores incluem o clônus e a hiper-reflexia. Os sinais neuronais motores inferiores incluem a atrofia, o enfraquecimento e as fasciculações. Para estabelecer o diagnóstico, é essencial: (1) exames de EMG mostrando desnervação ativa em 3 membros e na musculatura paraespinal; e (2) exames de condução nervosa normal que excluam o bloqueio de condução como causa de desnervação. Estes achados eletrofisiológicos, aliados à progressão inflexível dos sintomas e à exclusão de outros distúrbios que mimetizem as doenças neuronais motoras (ver adiante), solidificam o diagnóstico.
Diagnóstico diferencial
O diagnóstico diferencial da ELA inclui a síndrome pós-poliomielite em pacientes com episódio anterior de pólio paralítica, neuropatia motora multifocal com ou sem bloqueio de condução, endocrinopatias – em especial as condições de hiperparatireoide ou hipertireoide –, intoxicação com chumbo, infecções e síndromes paraneoplásicas.102,103
Tratamento
O curso da ELA é invariavelmente fatal. O objetivo primário da terapia consiste no tratamento dos sintomas.109,110 Para os pacientes com disfagia, a realização de uma gastrostomia endoscópica percutânea (GEP) deve ser considerada logo após a manifestação dos sintomas. A instituição de ventilação não invasiva deve ser planejada com cautela, equilibrando o risco de dependência da ventilação vs. o risco de morte súbita. Existem algoritmos disponíveis que destacam as indicações para instituição de GEP e assistência ventilatória.109,110 Fisioterapia, técnicas de reabilitação e dispositivos auxiliares (p. ex., bengalas, órtoses de tornozelo-pé e andadores) podem prolongar a independência funcional do paciente, contudo a maioria dos indivíduos afetados precisa usar cadeira de rodas. O riluzol é o único fármaco disponível para tratamento da ELA com uso aprovado pelo Food and Drug Administration (FDA). Acredita-se que este fármaco reduza a excitotoxicidade glutamato-mediada, embora seu modo de ação exato seja pouco conhecido. Estudos demonstraram que o riluzol retarda o declínio da força muscular e prolonga a sobrevida em até 4 meses.107 Este fármaco pode ser mais útil aos pacientes com doença branda ou em estágio inicial. Outras medicações são úteis para controlar a sialorreia, afeto pseudobulbar (labilidade emocional), dispneia e ansiedade.109,110 O encaminhamento do paciente para internação pode ser considerado durante a fase terminal.
O autor não possui relação comercial com os fabricantes de produtos e prestadores de serviços mencionados neste capítulo.
Referências
1. Cohn RD, Campbell KP. Molecular basis of muscular dystrophies. Muscle Nerve 2000;23:1456–71.
2. Manzur AY, Muntoni F. Diagnosis and new treatments in muscular dystrophies. J Neurol Neurosurg Psychiatry 2009;80:706–14.
3. Durbeej M, Campbell KP. Muscular dystrophies involving the dystrophin-glycoprotein complex: an overview of current mouse models. Curr Opin Genet Dev 2002;12:349– 61.
4. Beggs AH, Koenig M, Boyce FM, et al. Detection of 98% of DMD/BMD gene deletions by polymerase chain reaction. Hum Genet 1990;86:45–8.
5. Mathews KD. Muscular dystrophy overview: genetics and diagnosis. Neurol Clin 2003;21:795–816.
6. Muntoni F, Torelli S, Ferlini A. Dystrophin and mutations: one gene, several proteins, multiple phenotypes. Lancet Neurol 2003;2:731–40.
7. Mendell JR, Buzin CH, Feng J, et al. Diagnosis of Duchenne dystrophy by enhanced detection of small mutations. Neurology 2001;57:645–50.
8. Flanigan KM, von Niederhausern A, Dunn DM, et al. Rapid direct sequence analysis of the dystrophin gene. Am J Hum Genet 2003;72:931–9.
9. Finsterer J, Stollberger C. The heart in human dystrophinopathies. Cardiology 2003;99:1–19.
10. Bushby K, Finkel R, Birnkrant DJ, et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and pharmacological and psychosocial management. Lancet Neurol 2010;9:77–93.
11. Bonifati MD, Ruzza G, Bonometto P, et al. A multicenter, double-blind, randomized trial of defl azacort versus prednisone in Duchenne muscular dystrophy. Muscle Nerve 2000;23:1344–7.
12. Magali L, Lebacq J, Poortmans J, et al. Benefi cial effects of creatine supplementation in dystrophic patients. Muscle Nerve 2003;27:604–10.
13. Barton-Davis ER, Cordier L, Shoturma DI, et al. Aminoglycoside antibiotics restore dystrophin function to skeletal muscles of mdx mice. J Clin Invest 1999;104:367–8.
14. Wagner KR, Hamed S, Hadley DW, et al. Gentamicin treatment of Duchenne and Becker muscular dystrophy due to nonsense mutations. Ann Neurol 2001;49:706–11.
15. Gregorevic P, Chamberlain JS. Gene therapy for muscular dystrophy—a review of promising progress.Expert Opin Biol Ther 2003;3:803–14.
16. Fatkin D, MacRae C, Sasaki T, et al. Missense mutations in the rod domain of the lamin A/C gene as causes of dilated cardiomyopathy and conduction-system disease. N Engl J Med 1999;341:1715–24.
17. Rocha CT, Hoffman EP. Limb-girdle and congenital muscular dystrophies: current diagnostics, management, and emerging technologies. Curr Neurol Neurosci Rep 2010;10:267–76.
18. Guglieri M, Straub V, Bushby K, Lochmüller H. Limb-girdle muscular dystrophies. Curr Opin Neurol 2008;21: 576–84.
19. Minetti C, Sotgia F, Bruno C, et al. Mutations in the caveolin-3 gene cause autosomal dominant limb-girdle muscular dystrophy. Nat Genet 1998;18:365–8.
20. Nigro V. Molecular bases of autosomal recessive limb-girdle muscular dystrophies. Acta Myol 2003;22:35–42.
21. Selcen D, Engel AG. Mutations in myotilin cause myofi - brillar myopathy. Neurology 2004;62:1363–71.
22. Zatz M, de Paula F, Starling A, et al. The 10 autosomal recessive limb-girdle muscular dystrophies. Neuromuscul Disord 2003;13:532–44.
23. Betz RC, Schoser BG, Kasper D, et al. Mutations in CAV3 cause mechanical hyperirritability of skeletal muscle in rippling muscle disease. Nat Genet 2001;28:218–9.
24. Richard I, Broux O, Allamand V, et al. Mutations in the proteolytic enzyme calpain 3 cause limb-girdle muscular dystrophy type 2A. Cell 1995;81:27–40.
25. Liu J, Aoki M, Illa I, et al. Dysferlin, a novel skeletal muscle gene, is mutated in Miyoshi myopathy and limb girdle muscular dystrophy. Nat Genet 1998;20:31–6.
26. Bushby K. Diagnosis and management of the limb girdle muscular dystrophies. Pract Neurol 2009;9:314–23.
27. Duggan DJ, Gorospe JR, Fanin M, et al. Mutations in the sarcoglycan genes in patients with myopathy. N Engl J Med 1997;336:618–24.
28. Moreira ES, Wiltshire TJ, Faulkner G, et al. Limb-girdle muscular dystrophy type 2G is caused by mutations in the gene encoding the sarcomeric protein telethonin. Nat Genet 2000;24:163–6.
29. Poppe M, Cree L, Bourke J, et al. The phenotype of limb-girdle muscular dystrophy type 2I. Neurology 2003;60: 1246–51.
30. Frosk P, Greenberg CR, Tennese AA, et al. The most common mutation in FKRP causing limb girdle muscular dystrophy type 2I (LGMD2I) may have occurred only once and is present in Hutterites and other populations. Hum Mutat 2005;25:38–44.
31. Norwood F, de Visser M, Eymard B, et al. EFNS guideline on diagnosis and management of limb girdle muscular dystrophies. EFNS Guideline Task Force. Eur J Neurol 2007;14:1305–12.
32. Hackman P, Vihola A, Haravuori H, et al. Tibial muscular dystrophy is a titinopathy caused by mutations in TTN, the gene encoding the giant skeletal-muscle protein titin. Am J Hum Genet 2002;71:492–500.
33. Hewitt JE, Grewl PK. Glycosylation defects in inherited muscle disease. Cell Mol Life Sci 2003;60:251–8.
34. Grewal PK, Hewitt JE. Glycosylation defects: a new mechanism for muscular dystrophy. Hum Mol Genet 2003;12: R259–64.
35. Mendell JR, Boué DR, Martin PT. The congenital muscular dystrophies: recent advances and molecular insights. Pediatr Dev Pathol 2006;9:427–43.
36. Clement E, Mercuri E, Godfrey C, et al. Brain involvement in muscular dystrophies with defective dystroglycan glycosylation. Ann Neurol 2008;64:573–82.
37. Hayashi YK, Chou FL, Engvall E, et al. Mutations in the integrin alpha7 gene cause congenital myopathy. Nat Genet 1998;19:94–7.
38. Camacho Vanegas O, Bertini E, Zhang RZ, et al. Ullrich scleratonic muscular dystrophy is caused by recessive mutations in collagen type VI. Proc Natl Acad Sci U S A 2001;98:7516–21.
39. Flanigan KM, Kerr L, Bromberg MD, et al. Congenital muscular dystrophy with rigid spine syndrome: a clinical, pathological, radiological, and genetic study. Ann Neurol 2000;47:152–61.
40. Dalakas MC, Park KY, Semino-Mora C, et al. Desmin myopathy, a skeletal myopathy with cardiomyopathy caused by mutations in the desmin gene. N Engl J Med 2000;342:770–80.
41. Goldfarb LG, Vicart P, Goebel H, Dalakas MC. Desmin myopathy. Brain 2004;127:723–34.
42. Selcen D, Ohno K, Engel AG. Myofi brillar myopathy: clinical, morphological and genetic studies in 63 patients. Brain 2004;127:439–51.
43. Goldfarb JG, Dalakas MC. Tragedy in a heartbeat: malfunctioning desmin causes skeletal and cardiac muscle disease. J Clin Invest 2009;119:1806–13.
44. Banwell BL, Russel J, Fukudome T, et al. Myopathy, myasthenic syndrome, and epidermolysis bullosa simplex due to plectin defi ciency. J Neuropathol Exp Neurol 1999;58: 832–46.
45. Wheeler TM, Thornton CA. Myotonic dystrophy: RNA-mediated muscle disease. Curr Opin Neurol 2007;20: 572–6.
46. Day JW, Ricker K, Jacobsen JF, et al. Myotonic dystrophy type 2: molecular, diagnostic and clinical spectrum. Neurology 2003;60:657–64.
47. Kaliman P, Llagostera E. Myotonic dystrophy protein kinase (DMPK) and its role in the pathogenesis of myotonic dystrophy 1. Cell Signal 2008;20:1935–41.
48. Kanandia RN, Johnstone KA, Mankodi A, et al. A muscle-blind knockout model for myotonic dystrophy. Science 2003;302:1978–80.
49. Padberg GW, van Engelen BG. Facioscapulohumeral muscular dystrophy. Curr Opin Neurol 2009;22:539–42.
50. Rose MR, Tawil R. Drug treatment for facioscapulohumeral muscular dystrophy. Cochrane Database Syst Rev 2004;(2): CD002276.
51. Shanmugam V, Dion P, Rochefort D, et al. PABP2 polyalanine tract expansion causes intranuclear inclusions in oculopharyngeal muscular dystrophy. Ann Neurol 2000; 48:798–802.
52. Stowell KM. Malignant hyperthermia: a pharmacogenetic disorder. Pharmacogenomics 2008;9:1657–72.
53. Benca J, Hogan K. Malignant hyperthermia, coexisting disorders, and enzymopathies: risks and management options. Anesth Analg 2009;109:1049–53.
54. DiMauro S, Lamperti C. Muscle glycogenoses. Muscle Nerve 2001;24:984–99.
55. Hirano M, DiMauro S. Metabolic myopathies. Adv Neurol 2002;88:217–34.
56. Berardo A, DiMauro S, Hirano M. A diagnostic algorithm for metabolic myopathies. Curr Neurol Neurosci Rep 2010; 10:118–26.
57. Vissing J, Haller RG. The effect of oral sucrose on exercise tolerance in patients with McArdle’s disease. N Engl J Med 2003;349:2503–9.
58. Vasconcelos O, Sivakumar K, Dalakas MC, et al. Nonsense mutation in the human phosphofructokinase muscle subunit gene associated with retention of intron 10 in one of the isolated transcripts in Ashkenazi Jewish patients with Tarui disease. Proc Natl Acad Sci U S A 1995;92:10322–6.
59. Engel AG, Hirschhorn R, Huie ML. Acid maltase defi ciency. In: Engel AG, Franzini-Armstrong C, editors. Myology. New York: McGraw-Hill; 2004. p. 1559.
60. van der Ploeg AT, Clemens PR, Corzo D, et al. A randomized study of alglucosidase alfa in late-onset Pompe’s disease. N Engl J Med 2010;362:1396–406.
61. Dalakas MC, Leon-Monzon ME, Bernardini I, et al. The zidovudine-induced mitochondrial myopathy is associated with muscle carnitine defi ciency and lipid storage. Ann Neurol 1994;35:482–7.
62. Hassani A, Horvath R, Chinnery PF. Mitochondrial myopathies: developments in treatment. Curr Opin Neurol 2010 Jul 21. [Epub ahead of print]
63. DiMauro S, Schon EA. Mitochondrial respiratory-chain diseases. N Engl J Med 2003;348:2656–68.
64. McFarland R, Taylor RW, Turnbull DM. The neurology of mitochondrial DNA disease. Lancet Neurol 2002;1:343– 51.
65. Zeviani M, Carelli V. Mitochondrial disorders. Curr Opin Neurol 2003;16:585–94.
66. Meola G, Hanna MG, Fontaine B. Diagnosis and new treatment in muscle channelopathies. J Neurol Neurosurg Psychiatry 2009;80:360–5.
67. Celesia GG. Disorders of membrane channels or channelopathies. Clin Neurophysiol 2001;112:2–18.
68. Kleopa KA, Barchi RL. Genetic disorders of neuromuscular ion channels. Muscle Nerve 2002;26:299–325.
69. Raja Rayan DL, Hanna MG. Skeletal muscle channelopathies: nondystrophic myotonias and periodic paralysis. Curr Opin Neurol 2010 Jul 14. [Epub ahead of print]
70. Fouad G, Dalakas M, Servidei S, et al. Genotype-phenotype correlations of DHP receptor alpha 1-subunit gene mutations causing hypokalemic periodic paralysis. Neuromuscul Disord 1997;7:33–8.
71. Dalakas MC, Engel WK. Treatment of “permanent” muscle weakness in familial hypokalemic periodic paralysis. Muscle Nerve 1983;6:182–6.
72. Tawil R, McDermott MP, Brown R, et al. Randomized trial of dichlorphenamide in the periodic paralyses. Working Group on Periodic Paralysis. Ann Neurol 2000;47:46–53.
73. Farrugia ME, Vincent A. Autoimmune mediated neuromuscular junction defects. Curr Opin Neurol 2010 Jul 21. [Epub ahead of print]
74. McConville J, Vincent A. Diseases of the neuromuscular junction. Curr Opin Pharmacol 2002;2:296–301.
75. Sieb JP. Myopathies due to drugs, toxins, and nutritional defi ciency. In: Engel AG, Franzini-Armstrong C, editors. Myology. New York: McGraw-Hill; 2004. p. 1693.
76. Dalakas MC. Toxic and drug-induced myopathies. J Neurol Neurosurg Psychiatry 2009;80:832–8.
77. Dalakas MC. Infl ammatory and toxic myopathies. Curr Opin Neurol Neurosurg 1992;5:645–54.
78. Dalakas MC, Illa I, Pezeshkpour GH, et al. Mitochondrial myopathy caused by long-term zidovudine therapy. N Engl J Med 1990;322:1098–105.
79. Lewis W, Dalakas MC. Mitochondrial toxicity of antiviral drugs. Nat Med 1995;1:417–22.
80. Brinkman K, Smeitink JA, Romijn JA, et al. Mitochondrial toxicity induced by nucleoside-analogue reversetranscriptase inhibitors is a key factor in the pathogenesis of antiretroviral-therapy-related lipodystrophy. Lancet 1999;354:1112–5.
81. Rosenson RS. Current overview of statin-induced myopathy. Am J Med 2004;116:408–16.
82. Sieb JP, Gillessen T. Iatrogenic and toxic myopathies. Muscle Nerve 2003;27:142–56.
83. Kuncl RW. Colchicine myopathy and neuropathy. N Engl J Med 1987;316:1562–8.
84. Illa I, Dinsmore S, Dalakas MC. Immune-mediated mechanisms and immune activation of fi broblasts in the pathogenesis of eosinophilia-myalgia syndrome induced by L-tryptophan. Hum Pathol 1993;24:702–9.
85. Danon MJ, Carpenter S. Myopathy with thick fi lament loss following prolonged paralysis with vacuronium during steroid treatment. Muscle Nerve 1991;14:1131–9.
86. Bella I, Chad DA. Neuromuscular disorders and acute respiratory failure. Neurol Clin 1998;16:391–417.
87. Hirano M, Ott B, Raps E, et al. Acute quadriplegic myopathy: a complication of steroids, nondepolarizing blocking agents or both. Neurology 1992;42:2082–7.
88. Melli G, Chaudhry V, Cornblath DR. Rhabdomyolysis: an evaluation of 475 hospitalized patients. Medicine (Baltimore) 2005;84:377–85.
89. Hill M. The neuromuscular junction disorders. J Neurol Neurosurg Psychiatry 2003;74:ii32–ii37.
90. Leite MI, Waters P, Vincent A. Diagnostic use of autoantibodies in myasthenia gravis. Autoimmunity 2010 Apr 12. [Epub ahead of print] the Quality Standards Subcommittee of the American Academy of Neurology.
91. Aragones JM, Bolibar I, Bonfi ll X, et al. Myasthenia gravis: a higher than expected incidence in the elderly. Neurology 2003;60:1024–6.
92. Hoch W, McConville J, Helms S, et al. Auto-antibodies to the receptor tyrosine kinase MuSK in patients with myasthenia gravis without acetylcholine receptor antibodies. Nat Med 2001;7:365–8.
93. Sanders DB, El-Salem K, Massey JM. Clinical aspects of MuSK antibody positive seronegative MG. Neurology 2003;60:1978–80.
94. Vincent A, Bowen J, Newsom-Davis J. Seronegative generalised myasthenia gravis: clinical features, antibodies, and their targets. Lancet Neurol 2003;2:99–106.
95. Newsom-Davis J. Lambert-Eaton myasthenic syndrome. Curr Treat Options Neurol 2001;3:127–31.
96. Maddison P, Lang B, Mills K. Long-term outcome in Lambert-Eaton myasthenic syndrome without lung cancer. J Neurol Neurosurg Psychiatry 2001;70:212–7.
97. Sanders DB, Evoli A. Immunosuppressive therapies in myasthenia gravis. Autoimmunity 2010;43:428–35.
98. Wolfe GI, Kaminski HJ, Jaretzki A 3rd, et al. Development of a thymectomy trial in nonthymomatous myasthenia gravis patients receiving immunosuppressive therapy. Ann N Y Acad Sci 2003;998:473–80.
99. Hughes RA, Dalakas MC, Cornblath DR, et al. Clinical applications of intravenous immunoglobulins in neurology. Clin Exp Immunol 2009;158 Suppl 1:34–42.
100.Zebardast N, Patwa HS, Novella SP, Goldstein JM. Rituximab in the management of refractory myasthenia gravis. Muscle Nerve 2010;41:375–8.
101.Engel AG, Ohno K, Sine SM. Congenital myasthenic syndromes: a diverse array of molecular targets. J Neurocytol 2003;32:1017–37.
102.Nirmalananthan N, Greensmith L. Amyotrophic lateral sclerosis: recent advances and future therapies. Curr Opin Neurol 2005;18:712–9.
103.Brooks RB, Miller RG, Swash M, et al. El Escorial revisited: revised criteria for the diagnosis of ALS. World Federation of Neurology; 1998. Available at: http://www.wfnals. org/guidelines/1998elescorial/elescorial1998criteria.htm (accessed August 2010).
104.Kunst CB. Complex genetics of amyotrophic lateral sclerosis. Am J Hum Genet 2004;75:933–47.
105.Chen YZ, Bennet CL, Huynh HM, et al. DNA/RNA helicase gene mutations is a form of juvenile amyotrophic lateral sclerosis (AL4). Am J Hum Genet 2004;74:1128–35.
106.Cleveland DW, Rothstein JD. From Charcot to Lou Gehrig: deciphering selective motor neuron death in ALS. Nat Rev Neurosci 2001;2:806–19.
107.Bruijn LI, Miller M, Cleveland DW. Unraveling the mechanisms involved in motor neuron degeneration in ALS. Annu Rev Neurosci 2004;27:723–49.
108.Armon C. Smoking may be considered an established risk factor for sporadic ALS. Neurology 2009;73:1693–8.
109.Miller RG, Jackson CE, Kasarskis EJ, et al; Quality Standards Subcommittee of the American Academy of Neurology. Practice parameter update: the care of the patient with amyotrophic lateral sclerosis: multidisciplinary care, symptom management, and cognitive/behavioral impairment (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology.Neurology 2009;73:1227–33.
110.Simmons Z. Management strategies for patients with amyotrophic lateral sclerosis from diagnosis to death. Neurologist 2005;11:257–70.
FONTE: Marinos C. Dalakas, MD, FAAN
Professor of Neurology, Thomas Jefferson University Medical School, Philadelphia, PA
postado: luciano sousa
e-mail:lucianofisiol@gmail.com
facebook:lucianosousa lucianosousa
postado: luciano sousa
e-mail:lucianofisiol@gmail.com
facebook:lucianosousa lucianosousa